
Abstract
Epidemiological and clinical studies have suggested comorbidity between rheumatoid arthritis and Parkinson’s disease (PD), but whether there exists a causal association and the effect direction of rheumatoid arthritis on PD is controversial and elusive. To evaluate the causal relationship, we first estimated the genetic correlation between rheumatoid arthritis and PD, and then performed a two-sample Mendelian randomization analysis based on summary statistics from large genome-wide association studies of rheumatoid arthritis (N = 47,580) and PD (N = 482,703). We identified negative and significant correlation between rheumatoid arthritis and PD (genetic correlation: −0.10, P = 0.0033). Meanwhile, one standard deviation increase in rheumatoid arthritis risk was associated with a lower risk of PD (OR: 0.904, 95% CI: 0.866–0.943, P: 2.95E–06). The result was robust under all sensitivity analyses. Our results provide evidence supporting a protective role of rheumatoid arthritis on PD. A deeper understanding of the inflammation and immune response is likely to elucidate the potential pathogenesis of PD and identify therapeutic targets for PD.
Similar content being viewed by others
Parkinson’s disease (PD) is the second most common neurodegenerative disease, pathologically characterized by the progressive degeneration of dopaminergic neurons in the pars compacta of the substantia nigra1. The etiology of PD is multifarious and complex, with a sophisticated interaction between genetic and environmental factors2. Currently, compelling evidence implicates that inflammation and immunity play an important role in the pathogenesis of PD3, and the comorbidity between PD and autoimmune diseases has been reported4.
Rheumatoid arthritis is the most common autoimmune disorder, with a core feature of abnormal activation of inflammation and immunity. Although some cases of rheumatoid arthritis co-existing with PD have been reported4, the association between rheumatoid arthritis and PD remains elusive and controversial. Conventionally, rheumatoid arthritis was presumed to be associated with increased risk of PD, due to the overproduction of inflammatory mediators and overactivation of immune cells5,6. This hypothesis was further supported by studies from postmortem brain tissue of PD that demonstrated increased levels of proinflammatory mediators in the striatum of the brain7. However, recent cohort studies showed that patients with rheumatoid arthritis have a reduced risk of developing PD8,9, which was contrary to previous results. Nevertheless, such observational studies were influenced by the possibility of confounding factors, such as the use of nonsteroidal anti-inflammatory drugs (NSAIDs)8. The unadjusted confounding factors might bias the association evidence, as is difficult to avoid in observational studies. Therefore, whether rheumatoid arthritis affects the risk of PD remains unknown. Unraveling this association could help better understand the pathophysiology of PD and autoimmune diseases, and may facilitate the development of novel therapeutic targets.
Mendelian randomization (MR) is an advanced genetic epidemiological method, which is widely used to exploit the causal relationship between exposures and outcomes10,11. MR employs single nucleotide polymorphisms (SNPs) as instrumental variables for the exposure, thus it can effectively avoid the confounding factors from the change of related traits and has high scientific value. In order to decipher the potential causal relationship between rheumatoid arthritis and PD, a two-sample MR approach was employed to examine whether rheumatoid arthritis affects the risk of PD from a genetic perspective.
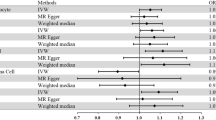
We first analyzed the genetic correlation between PD and rheumatoid arthritis, and detected a genome-wide significant and negative genetic correlation (genetic correlation: −0.10, P = 0.0033). Then we analyzed the role of rheumatoid arthritis on the risk of PD using the MR approach. As a result, each one standard deviation increase in rheumatoid arthritis risk was associated with a lower risk of PD (OR: 0.904, 95% CI: 0.866–0.943, P: 2.95E–06), and such results were supported using another three MR methods, namely MR–Egger regression (OR: 0.894, 95% CI: 0.836–0.957, P: 0.007), weighted mode (OR: 0.860, 95% CI: 0.805–0.918, P: 5.98E–04), and weighted median (OR: 0.915, 95% CI: 0.859–0.975, P: 0.006) (Fig. 1a, b). The funnel plot displays a symmetric pattern of effect size variation around the point estimate (Fig. 1c). The simple median method did not show a significant association but suggested the same direction of effect. The association was still significant after excluding variants within the MHC region, defined as base positions 24,000,000 to 35,000,000 on chromosome 6 (GRCh37) (OR: 0.920, 95% CI: 0.853–0.993, P: 0.031). We further replicated the results using exposures from another large GWAS on rheumatoid arthritis (OR: 0.929, 95% CI: 0.889–0.970, P: 8.60E–04) using the IVW method. And the association was significant after removing SNPs in the MHC region (OR: 0.930, 95% CI: 0.866–0.999, P: 0.047).

a Forest plot showing results from the Mendelian randomization (MR) analysis to evaluate potential causal associations between rheumatoid arthritis and PD. Estimates are per 1 standard deviation increase in the trait. b Scatter plot of single nucleotide polymorphism (SNP) potential effects on rheumatoid arthritis versus PD. The 95% CI for the effect size on PD is shown as vertical lines, while the 95% CI for the effect size on rheumatoid arthritis is shown as horizontal lines. The slope of fitted lines represents the estimated Mendelian randomization effect per method. c Funnel plot for rheumatoid arthritis shows the estimation using the inverse of the standard error of the causal estimate with each individual SNP as a tool. The vertical line represents the estimated causal effect obtained using IVW and MR–Egger method.
Furthermore, we performed extensive sensitivity analyses to validate the association between rheumatoid arthritis and PD. We did not detect heterogeneity of effects based on Cochran’s Q test results (Table 1). The F statistics of all the instrument variables were high enough (ranging from 27 to 1581) to support the absence of weakness in the chosen instrument variables. Meanwhile, no apparent horizontal pleiotropy was detected as the intercept of MR–Egger was not significantly deviated from zero (Table 1). Violation of the causality was not found by directionality examination using Steiger analysis either. And we detected no potential instrumental outlier at the nominal significance level of 0.05 with MR-PRESSO analysis. The leave-one-out results suggested that the estimated causal effect was not driven by a single instrumental variable.
In the past, various clinical studies have emerged to investigate the co-occurrence of rheumatoid arthritis and PD4. However, the relationship between rheumatoid arthritis and PD is still quite inconsistent and controversial6,8,9. For example, a population-based cohort study found that patients with rheumatoid arthritis had a significantly higher risk of developing PD (hazard ratio (HR): 1.14, 95% CI: 1.03–1.20)6. In contrast, another population-based case-control study from Denmark found that patients with rheumatoid arthritis have a decreased risk of developing PD (OR: 0.70, 95% CI: 0.60–0.90)8. Similar results were obtained from a retrospective cohort study involving 33,221 patients with newly diagnosed rheumatoid arthritis and 132,884 randomly selected age- and sex-matched patients without rheumatoid arthritis (HR: 0.65, 95% CI: 0.58–0.73)9. The heterogeneity of these results may be due to the inevitable confounding factors of observational studies. To clarify this relationship, we performed a two-sample MR analysis using genetic variants as instrumental variables, which can minimize the possibility of bias inherent to observational studies. As a result, our study provides novel and robust evidence for an inverse association between rheumatoid arthritis and the risk of PD.
Although our study provides evidence that patients with rheumatoid arthritis have a reduced risk of developing PD, the mechanisms underlying the protective effect is largely unknown. Our current finding together with earlier results8,9,12 contradicts the previous hypothesis that prolonged chronic inflammation and overproduction of inflammatory mediators in autoimmune diseases may activate microglia and contribute to the degeneration of neurons, thus increase the risk of developing PD8,13. The benefit of decreased risk for developing PD by rheumatoid arthritis cannot be attributed to therapeutic factors such as anti-inflammatory agent administration in rheumatoid arthritis either8, as MR approach elucidates the link using genetic instrumental variants. From another perspective, the activated microglia in the brain of PD patients may be a compensated consequence of the disease, and thus may be mainly associated with the progression of the disease, rather than initiation of the pathogenesis14. Further longitudinal observations of the progression rate of parkinsonism in patients with rheumatoid arthritis co-existing with PD may give credence to the viewpoint.
Notably, growing evidence implicates the role of lysosomal dysfunction in both autoimmune disorders and neurodegenerative diseases15. In the context of PD, the decrease of lysosomal enzyme activity, such as resulting from the mutation of GBA and LRRK22,16, may lead to the aggregation of α-synuclein and the formation of Lewy bodies, which is the pathological hallmark of PD17. Moreover, overexpression of lysosomal proteases cathepsin D could reduce the accumulation of α-synuclein aggregates in mice model18. Strikingly, elevated levels of lysosomal enzyme activity have been reported to occur in several autoimmune diseases, including rheumatoid arthritis15,19. In rheumatoid arthritis, high levels of several lysosomal cathepsins (B, D, G, K, L, and S) are present both in the serum and synovial fluid of patients, which is contradictory to the condition of PD with a lower level of lysosomal enzyme activity20. Therefore, rheumatoid arthritis may shed the protective effect on PD through the lysosome pathway, which is worth further exploration. In addition, all treatments available for PD are symptomatic only and could not slow down the progression up till now. Therefore, it’s urgent and important to search for new effective therapeutic approaches for PD. The present results suggested that explorations of the mechanisms of inflammation and immune response, as well as lysosomal dysfunction, have the potential to provide new targets for the treatment of PD.
There were also some limitations to the current study. We excluded SNPs that were associated with smoking and BMI, but there are still other potential confounders that could violate the independence assumption, and it’s not practicable to rule out all of them. Moreover, population stratification and potential sample overlap might be another source of bias, as in all MR analyses. However, the F statistics calculated in the sensitivity analysis were all large enough, suggesting that the bias might be minimal. Meanwhile, the GWAS used in the MR analysis was conducted based on participants of European ancestry, thus the findings might be biased and not applicable to other populations. Future studies in other non-European populations will provide a more comprehensive understandings. In addition, the effect size identified in our study was not that large as observed in previous cohort studies. Future exploration based on summary data from GWAS with a larger sample size was warranted to provide a more accurate estimate.
In conclusion, based on the MR analysis results obtained from large-scale GWAS summary data, we demonstrate that rheumatoid arthritis could reduce the risk of PD in the European-ancestry population. Further exploration of the molecular mechanisms underlying the crosstalk between peripheral and central immunity may shed light on the understanding of the protective effect of rheumatoid arthritis on PD and reveal therapeutic targets for PD.
Methods
Datasets
We obtained summarized association results for rheumatoid arthritis from genome-wide association study (GWAS) on rheumatoid arthritis of European ancestry (Ncase = 13,838, Ncontrol = 33,742)21. Details of the summary data were listed in Supplementary Table 1, and the study design like the collection of samples, quality control procedures and imputation methods have been described in the original publication. We selected genetic variants that passed the genome-wide significance threshold (P < 5E–08) as instrument variants. Since smoking and body mass index (BMI) are associated with risk of both rheumatoid arthritis and PD, we removed SNPs which have been reported to be suggestively associated with these two traits by GWAS searched in GWAS Catalog22 (P < 1E–05). Then we clumped the instrument variables based on the 1000 Genomes Project linkage disequilibrium (LD) structure, and kept index SNPs with the lowest P value (R2 < 0.001 with any other associated SNP within 10 Mb). The summary statistics for the retained instrument variables were shown in Supplementary Table 2. As a replication, we also extracted instrumental variables from another GWAS on rheumatoid arthritis of European ancestry (Ncase = 5539, Ncontrol = 20,169)23 using the method mentioned above. The detailed summary data was shown in Supplementary Table 3.
Summary statistics of PD risk for the selected instrument variables were obtained from the published PD GWAS of European ancestry with the largest sample size so far (Ncase = 33,647, Ncontrol = 449,056)24. The study design like the collection of samples, quality control procedures, and imputation methods have been described in the original publication. We carried out harmonization to rule out strand mismatches and ensure the SNP effect sizes on rheumatoid arthritis and PD correspond to the same allele. The summary statistics were shown in Supplementary Tables 2, 3. This study only utilized summarized results from published studies, and individual data were not involved.
Genetic correlation
We estimated the genetic correlation between PD and rheumatoid arthritis using GNOVA25 with default parameters. GNOVA estimates genetic covariance with the genetic variants summary data shared between two GWAS, and then calculates the genetic correlation based on genetic covariance and variant-based heritabilities. We ran GNOVA on all the SNPs in both diseases24,26 together with reference data derived from the 1000 Genomes Project European population.
Mendelian randomization analysis
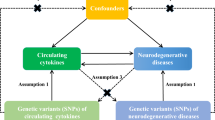
We hypothesized that rheumatoid arthritis as a risk factor could decrease the risk of PD, and used the MR approach to validate the hypothesis. The success of MR analysis depends on three assumptions: the genetic instruments are associated with risk of rheumatoid arthritis; the genetic instruments are not associated with potential confounders; the genetic instruments are associated with PD through the risk of rheumatoid arthritis (namely horizontal pleiotropy should not be present)20 (Supplementary Fig. 1).
We performed a two-sample MR analysis to estimate the effect of rheumatoid arthritis on PD by applying the fixed-effect inverse variance weighted (IVW) method, which is most widely used in MR studies and could provide robust causal estimates under the absence of directional pleiotropy. Meanwhile, we validated the results by applying MR–Egger regression, weighted mode, weighted median, and simple median methods in the discovery phase. We also performed MR analysis excluding variants within the extended major histocompatibility complex (MHC) region, since its complex LD structure made it susceptible to horizontal pleiotropy. In addition, to evaluate potential violations of the model assumptions in the MR analysis, we conducted extensive sensitivity analyses. Cochran’s Q test was computed to check heterogeneity across the individual causal effects. Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO) analysis was conducted to detect outlier instrument27. MR–Egger regression was performed to evaluate the directional pleiotropy of instruments. To evaluate the strongness of each instrument variable, we computed the F-statistic of each SNP as described by a previous study28. The leave-one-out analysis was conducted with the inverse variance weighted method to assess the influence of individual variants on the observed association. And we performed Steiger analysis29 to explore whether PD has a reverse causal impact on the risk of rheumatoid arthritis. The statistical power calculated at http://cnsgenomics.com/shiny/mRnd/ is sufficient (0.81) assuming the true causal OR of rheumatoid arthritis on PD is 0.9, given the involved sample size and the significance level α as 0.0530. The main statistical analyses were conducted using R package TwoSampleMR v0.5.531.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. Summary statistics of risk of PD was downloaded from IPDGC (https://pdgenetics.org/resources). Participants from 23andMe Inc. were excluded from the summary data.
Code availability
Code or algorithm used to generate results in this study are available from the corresponding authors on reasonable request. Exposure instrumental variables could be obtained using functions in R package TwoSampleMR.
References
Obeso, J. A. et al. Past, present, and future of Parkinson’s disease: a special essay on the 200th anniversary of the shaking palsy. Mov. Disord. 32, 1264–1310 (2017).
Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020).
Joshi, N. & Singh, S. Updates on immunity and inflammation in Parkinson disease pathology. J. Neurosci. Res. 96, 379–390 (2018).
Jiang, T., Li, G., Xu, J., Gao, S. & Chen, X. The Challenge of the Pathogenesis of Parkinson’s Disease: Is Autoimmunity the Culprit? Front. Immunol. 9, 2047 (2018).
Kunas, R. C., McRae, A., Kesselring, J. & Villiger, P. M. Antidopaminergic antibodies in a patient with a complex autoimmune disorder and rapidly progressing Parkinson’s disease. J. Allergy Clin. Immunol. 96, 688–690 (1995).
Chang, C. C. et al. Autoimmune rheumatic diseases and the risk of Parkinson disease: a nationwide population-based cohort study in Taiwan. Ann. Med. 50, 83–90 (2018).
Mogi, M. et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 180, 147–150 (1994).
Rugbjerg, K. et al. Autoimmune disease and risk for Parkinson disease: a population-based case-control study. Neurology 73, 1462–1468 (2009).
Sung, Y. F. et al. Reduced risk of Parkinson disease in patients with rheumatoid arthritis: a nationwide population-based study. Mayo Clin. Proc. 91, 1346–1353 (2016).
Lawlor, D. A., Harbord, R. M., Sterne, J. A., Timpson, N. & Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Li, C., Yang, W., Wei, Q. & Shang, H. Causal Association of Leukocytes Count and Amyotrophic Lateral Sclerosis: a Mendelian Randomization Study. Mol. Neurobiol. 57, 4622–4627 (2020).
Liu, F. C. et al. Inverse association of Parkinson disease with systemic lupus erythematosus: a nationwide population-based study. Medicine 94, e2097 (2015).
Perry, V. H. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav. Immun. 18, 407–413 (2004).
Fourgeaud, L. et al. TAM receptors regulate multiple features of microglial physiology. Nature 532, 240 (2016).
Bonam, S. R., Wang, F. & Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 18, 923–948 (2019).
Ysselstein, D. et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 10, 5570 (2019).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Cullen, V. et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain 2, 5 (2009).
Settembre, C., Fraldi, A., Medina, D. L. & Ballabio, A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 (2013).
Burgess, S. & Thompson, S. G. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol. 181, 251–260 (2015).
Eyre, S. et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 44, 1336–1340 (2012).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–d1012 (2019).
Stahl, E. A. et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 42, 508–514 (2010).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Lu, Q. et al. A powerful approach to estimating annotation-stratified genetic covariance via GWAS summary statistics. Am. J. Hum. Genet. 101, 939–964 (2017).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Burgess, S., Davies, N. M. & Thompson, S. G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 40, 597–608 (2016).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081 (2017).
Brion, M. J. A., Shakhbazov, K. & Visscher, P. M. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiol. 42, 1497–1501 (2013).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife https://doi.org/10.7554/eLife.34408 (2018).
Acknowledgements
This study was supported by the funding of the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC18038), the National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Grant No. Z20192006 and Z2018B08), the Science Foundation of Chengdu Science and Technology Bureau (2019-YF05-00307-SN), and the Sichuan Science and Technology Program (Grant No. 2021YJ0415).
Author information
Authors and Affiliations
Contributions
C.Y.L.: Conceptualization, methodology, analysis, and draft of the manuscript. R.W.O.: Revision of the manuscript. H.F.S.: Supervision, conceptualization, and revision of the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, C., Ou, R. & Shang, H. Rheumatoid arthritis decreases risk for Parkinson’s disease: a Mendelian randomization study. npj Parkinsons Dis. 7, 17 (2021). https://doi.org/10.1038/s41531-021-00166-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-021-00166-x
This article is cited by
-
Causal relationships between type 1 diabetes mellitus and Alzheimer’s disease and Parkinson’s disease: a bidirectional two-sample Mendelian randomization study
European Journal of Medical Research (2024)
-
Genome-wide analyses identify NEAT1 as genetic modifier of age at onset of amyotrophic lateral sclerosis
Molecular Neurodegeneration (2023)
-
Bidirectional Mendelian randomization analysis of the genetic association between primary lung cancer and colorectal cancer
Journal of Translational Medicine (2023)
-
Innate and adaptive immune abnormalities underlying autoimmune diseases: the genetic connections
Science China Life Sciences (2023)
