
Abstract
Duchenne muscular dystrophy (DMD) is a muscle wasting disorder caused by mutations in the gene encoding dystrophin. Gene therapy using micro-dystrophin (MD) transgenes and recombinant adeno-associated virus (rAAV) vectors hold great promise. To overcome the limited packaging capacity of rAAV vectors, most MD do not include dystrophin carboxy-terminal (CT) domain. Yet, the CT domain is known to recruit α1- and β1-syntrophins and α-dystrobrevin, a part of the dystrophin-associated protein complex (DAPC), which is a signaling and structural mediator of muscle cells. In this study, we explored the impact of inclusion of the dystrophin CT domain on ΔR4-23/ΔCT MD (MD1), in DMDmdx rats, which allows for relevant evaluations at muscular and cardiac levels. We showed by LC-MS/MS that MD1 expression is sufficient to restore the interactions at a physiological level of most DAPC partners in skeletal and cardiac muscles, and that inclusion of the CT domain increases the recruitment of some DAPC partners at supra-physiological levels. In parallel, we demonstrated that inclusion of the CT domain does not improve MD1 therapeutic efficacy on DMD muscle and cardiac pathologies. Our work highlights new evidences of the therapeutic potential of MD1 and strengthens the relevance of this candidate for gene therapy of DMD.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others
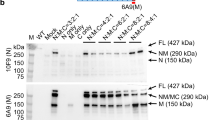

Data availability
The data that support the findings of this study are available from the corresponding authors on reasonable request.
References
Mendell JR, Lloyd-Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve. 2013;48:21–6.
Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28.
Bhat HF, Mir SS, Dar KB, Bhat ZF, Shah RA, Ganai NA. ABC of multifaceted dystrophin glycoprotein complex (DGC). J Cell Physiol. 2018;233:5142–59.
Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–9.
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93.
Wang D, Zhong L, Nahid MA, Gao G. The potential of adeno-associated viral vectors for gene delivery to muscle tissue. Expert Opin Drug Deliv. 2014;11:345–64.
Rivera VM, Gao G, Grant RL, Schnell MA, Zoltick PW, Rozamus LW, et al. Long-term pharmacologically regulated expression of erythropoietin in primates following AAV-mediated gene transfer. Blood. 2005;105:1424–30.
Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther Janv. 2010;18:80–6.
Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–61.
Athanasopoulos T, Graham I, Foster H, Dickson G. Recombinant adeno-associated viral (rAAV) vectors as therapeutic tools for Duchenne muscular dystrophy (DMD). Gene Ther. 2004;11:S109–21.
Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–40.
Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787–9.
Foster H, Sharp PS, Athanasopoulos T, Trollet C, Graham IR, Foster K, et al. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 Gene Transfer. Mol Ther. 2008;16:1825–32.
Bostick B, Yue Y, Lai Y, Long C, Li D, Duan D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum Gene Ther. 2008;19:851–6.
Yue Y, Pan X, Hakim CH, Kodippili K, Zhang K, Shin J-H, et al. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum Mol Genet. 2015;24:5880–90.
Le Guiner C, Servais L, Montus M, Larcher T, Fraysse B, Moullec S, et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat Commun. 2017;8:16105.
Hakim CH, Wasala NB, Pan X, Kodippili K, Yue Y, Zhang K, et al. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of duchenne muscular dystrophy. Mol Ther Methods Clin Dev. 2017;6:216–30.
Duan D, Systemic AAV. Micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol Ther. 2018;26:2337–56.
Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–35.
Gregorevic P, Blankinship MJ, Allen JM, Chamberlain JS. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol Ther. 2008;16:657–64.
Shin J-H, Nitahara-Kasahara Y, Hayashita-Kinoh H, Ohshima-Hosoyama S, Kinoshita K, Chiyo T. et al. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011;18:910–9.
Bostick B, Shin J-H, Yue Y, Duan D. AAV-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol Ther. 2011;19:1826–32.
Wang B, Li J, Fu FH, Xiao X. Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J Orthopaedic Res. 2009;27:421–6.
Guiner CL, McIntyre M, Larcher T, Adjali O, Lafoux A, Toumaniantz G, et al. Dose finding study in the DMDmdx rat model to determine the efficacious dose of a rAAV9 vector encoding a human mini-dystrophin after IV administration. Neuromuscular Disorders. 2017;27:S188.
Mendell JR, Sahenk Z, Lehman K, Nease C, Lowes LP, Miller NF, et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020;77:1122–31.
Moorehead T, Yong F, Neelakantan S, Beaverson K, Binks M. Safety and tolerability of PF-06939926 in ambulatory boys with duchenne muscular dystrophy: a phase 1b multicenter, open- label, dose ascending study. Mol Ther J Am Soc Gene Ther. 2020;28:272.
Koo T. Studies on gene transfer in skeletal muscle cells and tissues using recombinant adeno-associated virus (AAV) vectors. Thesis. 2010.
Suzuki A, Yoshida M, Ozawa E. Mammalian alpha 1- and beta 1-syntrophin bind to the alternative splice-prone region of the dystrophin COOH terminus. J Cell Biol. 1995;128:373–81.
Sadoulet-Puccio HM, Rajala M, Kunkel LM. Dystrobrevin and dystrophin: an interaction through coiled-coil motifs. Proc Natl Acad Sci USA. 1997;94:12413–8.
Yoshida M, Hama H, Ishikawa-Sakurai M, Imamura M, Mizuno Y, Araishi K, et al. Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Hum Mol Genet. 2000;9:1033–40.
Bhat HF, Adams ME, Khanday FA. Syntrophin proteins as Santa Claus: role(s) in cell signal transduction. Cell Mol Life Sci. 2013;70:2533–54.
Matamoros M, Pérez-Hernández M, Guerrero-Serna G, Amorós I, Barana A, Núñez M, et al. Nav1.5 N-terminal domain binding to α1-syntrophin increases membrane density of human Kir2.1, Kir2.2 and Nav1.5 channels. Cardiovasc Res. 2016;110:279–90.
Leyva-Leyva M, Sandoval A, Felix R, González-Ramírez R. Biochemical and functional interplay between ion channels and the components of the dystrophin-associated glycoprotein complex. J Membr Biol. 2018;251:535–50.
Sabourin J, Lamiche C, Vandebrouck A, Magaud C, Rivet J, Cognard C, et al. Regulation of TRPC1 and TRPC4 cation channels requires an α1-syntrophin-dependent complex in skeletal mouse myotubes. J Biol Chem. 2009;284:36248–61.
Vandebrouck A, Sabourin J, Rivet J, Balghi H, Sebille S, Kitzis A, et al. Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. 2007;21:608–17.
Dombernowsky NW, Ölmestig JNE, Witting N, Kruuse C. Role of neuronal nitric oxide synthase (nNOS) in Duchenne and Becker muscular dystrophies - Still a possible treatment modality? Neuromuscul Disord. 2018;28:914–26.
Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin cooh-terminal domain. J Cell Biol. 2000;150:1399–410.
Koo T, Malerba A, Athanasopoulos T, Trollet C, Boldrin L, Ferry A, et al. Delivery of AAV2/9-microdystrophin genes incorporating helix 1 of the coiled-coil motif in the C-Terminal domain of dystrophin improves muscle pathology and restores the level of α1-syntrophin and α-dystrobrevin in skeletal muscles of mdx mice. Hum Gene Ther. 2011;22:1379–88.
McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech. 2015;8:195–213.
Tandon A, Jefferies JL, Villa CR, Hor KN, Wong BL, Ware SM, et al. Dystrophin genotype-cardiac phenotype correlations in Duchenne and Becker muscular dystrophies using cardiac magnetic resonance imaging. Am J Cardiol. 2015;115:967–71.
Johnson EK, Zhang L, Adams ME, Phillips A, Freitas MA, Froehner SC, et al. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE. 2012;7:e43515.
Larcher T, Lafoux A, Tesson L, Remy S, Thepenier V, François V, et al. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS ONE. 2014;9:e110371.
Li X, Eastman EM, Schwartz RJ, Draghia-Akli R. Synthetic muscle promoters: activities exceeding naturally occurring regulatory sequences. Nat Biotechnol. 1999;17:241–5.
D’Costa S, Blouin V, Broucque F, Penaud-Budloo M, François A, Perez IC, et al. Practical utilization of recombinant AAV vector reference standards: focus on vector genomes titration by free ITR qPCR. Mol Ther Methods Clin Dev. 2016;5:16019.
Salvetti A, Orève S, Chadeuf G, Favre D, Cherel Y, Champion-Arnaud P, et al. Factors influencing recombinant adeno-associated virus production. Hum Gene Ther. 1998;9:695–706.
Shinoda K, Tomita M, Ishihama Y. emPAI Calc—for the estimation of protein abundance from large-scale identification data by liquid chromatography-tandem mass spectrometry. Bioinformatics. 2010;26:576–7.
Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–72.
Fraysse B, Desaphy J-F, Rolland J-F, Pierno S, Liantonio A, Giannuzzi V, et al. Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol Dis. 2006;21:372–80.
Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol. 2011;51:288–98.
Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50.
Dyle MC, Ebert SM, Cook DP, Kunkel SD, Fox DK, Bongers KS, et al. Systems-based discovery of tomatidine as a natural small molecule inhibitor of skeletal muscle atrophy. J Biol Chem. 2014;289:14913–24.
Moorwood C, Liu M, Tian Z, Barton ER. Isometric and eccentric force generation assessment of skeletal muscles isolated from murine models of muscular dystrophies. J Vis Exp. 2013;31:e50036.
Chapdelaine P, Gérard C, Sanchez N, Cherif K, Rousseau J, Ouellet DL, et al. Development of an AAV9 coding for a 3XFLAG-TALEfrat#8-VP64 able to increase in vivo the human frataxin in YG8R mice. Gene Ther. 2016;23:606–14.
Mayra A, Tomimitsu H, Kubodera T, Kobayashi M, Piao W, Sunaga F, et al. Intraperitoneal AAV9-shRNA inhibits target expression in neonatal skeletal and cardiac muscles. Biochem Biophys Res Commun. 2011;405:204–9.
Madhavan R, Jarrett HW. Phosphorylation of dystrophin and alpha-syntrophin by Ca(2+)-calmodulin dependent protein kinase II. Biochim Biophys Acta. 1999;1434:260–74.
Madhavan R, Jarrett HW. Calmodulin-activated phosphorylation of dystrophin. Biochemistry. 1994;33:5797–804.
Sato S, Omori Y, Katoh K, Kondo M, Kanagawa M, Miyata K, et al. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat Neurosci. 2008;11:923–31.
Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, et al. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–9.
Banks GB, Judge LM, Allen JM, Chamberlain JS. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6:e1000958.
Lorin C, Vögeli I, Niggli E. Dystrophic cardiomyopathy: role of TRPV2 channels in stretch-induced cell damage. Cardiovasc Res. 2015;106:153–62.
Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–30.
Hermans MCE, Pinto YM, Merkies ISJ, de Die-Smulders CEM, Crijns HJGM, Faber CG. Hereditary muscular dystrophies and the heart. Neuromuscul Disord. 2010;20:479–92.
Szabó PL, Ebner J, Koenig X, Hamza O, Watzinger S, Trojanek S, et al. Cardiovascular phenotype of the Dmdmdx rat - a suitable animal model for Duchenne muscular dystrophy. Dis Model Mech. 2021;22:14.
England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343:180–2.
Yue Y, Liu M, Duan D. C-Terminal-Truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double-knockout mice. Mol Ther. 2006;14:79–87.
Nakamori M, Takahashi MP. The role of α-dystrobrevin in striated muscle. Int J Mol Sci. 2011;12:1660–71.
Ishikawa-Sakurai M, Yoshida M, Imamura M, Davies KE, Ozawa EZZ. domain is essentially required for the physiological binding of dystrophin and utrophin to beta-dystroglycan. Hum Mol Genet. 2004;13:693–702.
Johnson EK, Li B, Yoon JH, Flanigan KM, Martin PT, Ervasti J, et al. Identification of new dystroglycan complexes in skeletal muscle. PLoS ONE. 2013;8:e73224.
Liu L. Lessons from cavin-1 deficiency. Biochem Soc Trans. 2020;48:147–54.
Taniguchi T, Maruyama N, Ogata T, Kasahara T, Nakanishi N, Miyagawa K, et al. PTRF/Cavin-1 deficiency causes cardiac dysfunction accompanied by cardiomyocyte hypertrophy and cardiac fibrosis. PLoS ONE. 2016;11:e0162513.
Kaakinen M, Reichelt ME, Ma Z, Ferguson C, Martel N, Porrello ER, et al. Cavin-1 deficiency modifies myocardial and coronary function, stretch responses and ischaemic tolerance: roles of NOS over-activity. Basic Res Cardiol. 2017;112:24.
Bostick B, Yue Y, Long C, Marschalk N, Fine DM, Chen J, et al. Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged mdx mice. Mol Ther. 2009;17:253–61.
Banks GB, Combs AC, Chamberlain JR, Chamberlain JS. Molecular and cellular adaptations to chronic myotendinous strain injury in mdx mice expressing a truncated dystrophin. Hum Mol Genet. 2008;17:3975–86.
Banks GB, Chamberlain JS, Froehner SC. Truncated dystrophins can influence neuromuscular synapse structure. Mol Cell Neurosci. 2009;40:433–41.
Sawicka E. Origin of the ring muscle fibers in neuromuscular diseases. Neuropatologia Polska. 1991;29:29–40.
Sekiguchi M. The role of dystrophin in the central nervous system: a mini review. Acta Myol: Myopathies and Cardiomyopathies: Official Journal of the Mediterranean Society of Myology. 2005;24:93–7.
Haenggi T, Fritschy J-M. Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue. Cell Mol Life Sci: CMLS. 2006;63:1614–31.
Chen L, Zhang J, Hu X, Philipson KD, Scharf SM. The Na+/Ca2+ exchanger-1 mediates left ventricular dysfunction in mice with chronic intermittent hypoxia. J Appl Physiol (1985). 2010;109:1675–85.
Acknowledgements
We thank all the personnel of the Boisbonne Center for Gene Therapy (ONIRIS, INSERM, Nantes, France) and of the UTE IRS2 (University of Nantes, France) for the handling and care of the rats included in this study. We also thank the vector core of UMR 1089 (CPV, INSERM and University of Nantes) for the cloning of the MDs pAAV plasmids and the production of the rAAV vectors used in this study. We thank the Mass Spectrometry and Proteomics Facility (Ohio State University, Columbus, Ohio, USA), which performed the proteomics experiments. We thank E. Marrosu (UCL Great Ormond Street Institute of Child Health, London, United Kingdom) for providing MANEX1011B and MW8 antibodies for co-immunoprecipitation experiments. We thank G. Potier for his valuable help during the analyses of label free quantitation. We also thank T. Cronin for editing the English language of this manuscript. Finally, we thank the MDA Monoclonal Antibody Resource for providing the MANEX 1011 C antibody.
Funding
This project was supported by the MDA (Muscular Dystrophy Association, Research Grant ID #513878), the AFM-Téléthon (Association Française contre les Myopathies), the “Fondation d’entreprise pour la thérapie génique en Pays de la Loire”, INSERM, INRA, the University of Nantes and the University Hospital of Nantes. The Fusion Orbitrap instrument used for proteomics study was supported by NIH Award Number Grant S10 OD018056.
Author information
Authors and Affiliations
Contributions
Study design and reviewing of the data: AB, VF, FM, CLG. Experimental investigation: AB, LZ, AL, BF, GT, TL, TG, ML, CL, AH, AC. Resources: VF, MA, BM, JG, VB, SR, IA, CH, AM, BK, ALH, LP and FM. Writing – original draft: AB, LZ, AL, BF and CLG. Writing – Review & Editing: all authors. Supervision: CLG and OA. Funding acquisition: CLG, PM, GD and OA.
Corresponding author
Ethics declarations
Competing interests
PM, GD and CLG are co-authors of a patent for systemic treatment of dystrophic pathologies (EP3044319A1, dated 27 June 2014). GD is an inventor on a PCT for production of large-sized micro-dystrophins in an AAV-based vector configuration (PCT/EP2016/060350, dated 17 May 2015). The remaining authors declare no competing financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bourdon, A., François, V., Zhang, L. et al. Evaluation of the dystrophin carboxy-terminal domain for micro-dystrophin gene therapy in cardiac and skeletal muscles in the DMDmdx rat model. Gene Ther 29, 520–535 (2022). https://doi.org/10.1038/s41434-022-00317-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-022-00317-6
This article is cited by
-
Therapeutic approaches for Duchenne muscular dystrophy
Nature Reviews Drug Discovery (2023)
-
Micro-dystrophin gene constructs for repairing heart and muscle function in rats: the smaller is enough?
Gene Therapy (2022)
