
Abstract
In female mammals, the proliferation, apoptosis, and estradiol-17β (E2) secretion of granulosa cells (GCs) have come to decide the fate of follicles. DNA methylation and RSPO2 gene of Wnt signaling pathway have been reported to involve in the survival of GCs and follicular development. However, the molecular mechanisms for how DNA methylation regulates the expression of RSPO2 and participates in the follicular development are not clear. In this study, we found that the mRNA and protein levels of RSPO2 significantly increased during follicular development, but the DNA methylation level of RSPO2 promoter decreased gradually. Inhibition of DNA methylation or DNMT1 knockdown could decrease the methylation level of CpG island (CGI) in RSPO2 promoter and upregulate the expression level of RSPO2 in porcine GCs. The hypomethylation of −758/−749 and −563/−553 regions in RSPO2 promoter facilitated the occupancy of transcription factor E2F1 and promoted the transcriptional activity of RSPO2. Moreover, RSPO2 promoted the proliferation of GCs with increasing the expression level of PCNA, CDK1, and CCND1 and promoted the E2 secretion of GCs with increasing the expression level of CYP19A1 and HSD17B1 and inhibited the apoptosis of GCs with decreasing the expression level of Caspase3, cleaved Caspase3, cleaved Caspase8, cleaved Caspase9, cleaved PARP, and BAX. In addition, RSPO2 knockdown promoted the apoptosis of GCs, blocked the development of follicles, and delayed the onset of puberty with decreasing the expression level of Wnt signaling pathway-related genes (LGR4 and CTNNB1) in vivo. Taken together, the hypomethylation of −758/−749 and −563/−553 regions in RSPO2 promoter facilitated the occupancy of E2F1 and enhanced the transcription of RSPO2, which further promoted the proliferation and E2 secretion of GCs, inhibited the apoptosis of GCs, and ultimately ameliorated the development of follicles through Wnt signaling pathway. This study will provide useful information for further exploration on DNA-methylation-mediated RSPO2 pathway during follicular development.
Similar content being viewed by others


Introduction
In female mammals, the normal follicular development and ovarian maturation are essential for the initiation of puberty and acquisition of the capacity to fertilization and reproduction [1, 2]. In mice, knocking out the KISS1 gene, which is well known for the gate regulator of pubertal timing, resulted in the absence of mature follicles and failure of pubertal transition [3]. Previous studies have shown that the proliferation and apoptosis of granulosa cells (GCs) as well as the synthesis of estradiol-17β (E2) are required for the follicular development [4,5,6]. The excessive apoptosis and death of GCs is quite possible to directly or indirectly block follicular development [7, 8]. For example, the disruption of estrogen receptor beta expression in mutant mice reduces the E2 production of GCs [9, 10], increases the apoptosis of GCs, and results in the failure of follicular maturation [11] and ovulatory dysfunction [12, 13]. In premature ovarian failure mice, upregulation of anti-Müllerian hormone expression in GCs inhibits the apoptosis of GCs and promotes the follicular development [14]. These observations suggest that the proliferation, apoptosis, and secretion of E2 in GCs have come to decide the fate of follicles in mammals. However, the mechanisms by which the GCs regulate the follicular development and pubertal initiation have not been fully understood.
Accumulating studies demonstrate the crucial roles of DNA methylation in the follicular development and the onset of puberty [15, 16]. DNA methylation is adding the methyl on the cytosine residues to regulate the transcription of genes mainly by DNA methyltransferases 1 (DNMT1) [17]. Previous studies have shown that the disrupted DNA methylation contributes to the arrest of follicular development and delay of puberty in rats [18, 19]. DNMT1-mediated MEG3 hypermethylation has been related to puberty by inhibiting the P53 signaling pathway [20, 21]. In polycystic ovary syndrome (PCOS) patients, DNMT1-dependent CDKN1A promoter hypomethylation inhibits the proliferation and growth of GCs [22]. In bovine luteal cells, the hypermethylation of the CYP19A1 gene promoter inhibits its expression to suppress the production of E2 [23, 24]. In rats, the hypomethylation of Caspase3 promoter promotes its expression and leads to the apoptosis of GCs [25]. LINE1 CpG-DNA hypomethylation in women GCs is strongly associated with PCOS [26]. In cumulus GCs of women with PCOS, the hypermethylation of TNF inhibits its expression to hinder cumulus oocyte complex expansion and compromise ovulation [27]. In summary, the change of DNA methylation in GCs may participate in follicular development and pubertal initiation by regulating the expression of key genes.
The RSPO2 gene is a member of the R-Spondin family, which activates and strongly potentiates the Wnt signaling pathway through their homologous LGR4, LGR5, and LGR6 receptors [28,29,30]. In mammals, RSPO family plays an important role in ovarian cell proliferation, apoptosis, and follicular development [31,32,33]. For example, interfering with the transcription of RSPO1 inhibits the cellular proliferation and promotes cellular apoptosis in human ovarian cancer cells [31]. RSPO2 promotes the transformation of primary follicles into secondary follicles in mouse ovaries [32]. Knockout RSPO2 inhibits the proliferation and differentiation of mouse ovarian GCs and impedes the growth of follicles [33]. In this study, we found that two CpG islands (CGIs) existed in the promoter of RSPO2. Therefore, we hypothesized that DNA methylation might be involved in the proliferation, apoptosis, and E2 secretion of porcine GCs by mediating the expression of RSPO2, thus affecting the follicular development and onset of puberty in mammals.
Results
DNA hypomethylation in CGI2 promoted the expression level of RSPO2 during follicular development
In order to explore whether DNA methylation was involved in the expression of RSPO2 during follicular development, the methylation and expression levels of RSPO2 were detected. There were two CGIs in the promoter of RSPO2 (CGI1, 217 base pair (bp), −1437/−1220 bp and CGI2, 669 bp, −1078/−409 bp, transcription start site = +1; Fig. 1A). During the development of porcine follicles, the DNA methylation levels of CGI2-1 decreased gradually across small follicles (1–3 mm), medium follicles (3–5 mm), and large follicles (5–7 mm) (Fig. 1B), but the mRNA and protein expression levels of RSPO2 significantly increased (Fig. 1C). To further confirm the regulation of DNA methylation on the expression of RSPO2, the expression and methylation levels of RSPO2 in GCs were examined after treating with DNA methylation inhibitor (5-Aza-CDR). We found that inhibition of DNA methylation significantly upregulated the mRNA and protein expression levels of RSPO2 in GCs (Fig. 1D) and reduced the methylation levels of non-CpG islands (NCGIs) and CGIs at RSPO2 promoter (Fig. 1E). Furthermore, to identify the core regulatory region of DNA-methylation-mediated RSPO2 expression, we segmented and methylated the NCGIs (pGL3-NCGI1: −1540/−1357 bp, pGL3-NCGI2: −1187/−1003 bp, and pGL3-NCGI3: −327/−76 bp) and CGIs (pGL3-CGI1: −1345/−1165 bp, pGL3-CGI2-1: −1014/−721 bp, and pGL3-CGI2–2: −709/−362 bp) of RSPO2 promoter. Dual-luciferase activity analysis showed that the methylated pGL3-CGI2-1 and pGL3-CGI2–2 significantly reduced the transcriptional activity of RSPO2, compared with the unmethylated pGL3-CGI2-1 and pGL3-CGI2–2. But the methylated pGL3-NCGI1, pGL3-CGI1, pGL3-NCGI2, and pGL3-NCGI3 did not exhibit marked effect on the transcriptional activity of RSPO2 (Fig. 1F). These results indicated that the hypomethylation in CGI2 promoted the expression of RSPO2 during follicular development.

A Schematic distribution of the CpG islands and bisulfite sequencing primers in the promoter of RSPO2. Blue region is the CpG island; red and horizontal bars show the positions of bisulfite primers. B Bisulfite sequencing was conducted to determine the methylation status of CGI2-1 at the RSPO2 promoter in small (1–3 mm), medium (3–5 mm), and large (5–7 mm) follicles. C The mRNA and protein expression level of RSPO2 in small, medium, and large follicles. D The mRNA and protein expression level of RSPO2 in porcine GCs treated with 5-Aza-CdR. The numbers below bands were folds of band intensities relative to control. The band intensities were measured by ImageJ and normalized to Tubulin. E BSP was used to detect the NCGI and CGI methylation status of RSPO2 in porcine GCs treated with 5-Aza-CdR. F The luciferase reporter plasmids carrying different regions of RSPO2 were methylated in vitro and transfected into GCs. Relative luciferase activity in GCs were determined. *P < 0.05, **P < 0.01. CGI CpG island, NCGI non-CpG island.
RSPO2 promoted the proliferation of porcine ovarian GCs
To determine the effect of RSPO2 on the proliferation of GCs, RSPO2 overexpression vector (pcDNA3.1-RSPO2) or RSPO2 small interfering RNA (RSPO2-siRNA) were transfected into GCs, and the proliferation ability and expression of genes related to proliferation pathways were detected. We initially confirmed the efficiency of RSPO2 overexpression and knockdown through quantitative reverse transcription PCR (qRT-PCR) and western blotting (WB). The mRNA and protein expression levels of RSPO2 in GCs increased significantly after transfecting with pcDNA3.1-RSPO2 (Fig. 2A), and the overexpression effect of RSPO2 increased with the increase of vector concentration and 500 ng of pcDNA3.1-RSPO2 was selected for subsequent experiments as it exhibited the strongest overexpression efficiency. Three RSPO2-siRNAs (RSPO2-siRNA1, RSPO2-siRNA2, and RSPO2-siRNA3) were synthesized and transfected into GCs for RSPO2 knockdown. RSPO2-siRNA3 significantly reduced the mRNA and protein expression levels of RSPO2 (Fig. 2B) and was selected for subsequent experiments as it exhibited the strongest knockdown efficiency. The qRT-PCR (Fig. 2C) and WB (Fig. 2D) showed that RSPO2 overexpression significantly promoted the mRNA expression level of cell cycle-related genes (PCNA, CDK1, CDK2, CCNA1, CCND1, and CCNE2) as well as the protein expression of PCNA, CDK1, and CCND1. Moreover, 5-ethynyl-2′-deoxyuridine (EdU) staining (Fig. 2E) and 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay (Fig. 2F) showed that RSPO2 overexpression significantly increased the proliferation ability of GCs. Meanwhile, RSPO2 silencing significantly inhibited the mRNA expression level of PCNA, CDK1, CCNA1, and CCNE2 as well as the protein expression of PCNA, CDK1, and CCND1 (Fig. 2G, H) and significantly inhibited the proliferation of GCs (Fig. 2I, J). Furthermore, we synthesized and transfected GCs with RSPO2-siRNA4 and RSPO2-siRNA5 to avoid off-target effect-related phenotype. Based on the knockdown efficiency, RSPO2-siRNA5 was selected and significantly reduced the mRNA and protein expression levels of RSPO2 (Fig. 2K) and inhibited the mRNA expression of PCNA, CDK1, CCNA1, and CCNB1 (Fig. 2L). EdU staining (Fig. 2M) and MTT assay (Fig. 2N) demonstrated that RSPO2 knockdown by RSPO2-siRNA5 significantly depressed the proliferation of GCs. Taken together, these results suggested that RSPO2 promoted the proliferation of porcine ovarian GCs.

A, B The mRNA and protein expression levels of RSPO2 after transfection with pcDNA3.1-RSPO2 (A) or RSPO2-siRNA (B) in porcine GCs. The numbers below bands were folds of band intensities relative to control. The band intensities were measured by ImageJ and normalized to Tubulin. C, D The mRNA (C) and protein (D) expression levels of PCNA, CDK1, and CCND1 in porcine GCs with RSPO2 overexpression. E, F The proliferation rates of GCs with RSPO2 overexpression were assessed by EdU (E) and MTT assay (F). G, H The mRNA (G) and protein (H) expression levels of PCNA, CDK1, and CCND1 in porcine GCs with RSPO2 knockdown. I, J The proliferation rates of GCs with RSPO2 knockdown were assessed by EdU (I) and MTT assay (J). K The mRNA and protein expression levels of RSPO2 after transfection with another two RSPO2-siRNAs (RSPO2-siRNA4 and 5) in porcine GCs. L The mRNA expression levels of cell cycle-related genes in porcine GCs transfected with RSPO2-siRNA5. M, N The proliferation rates of GCs transfected with RSPO2-siRNA5 was assessed by EdU (M) and MTT assay (N). *P < 0.05, **P < 0.01.
RSPO2 inhibited the apoptosis of porcine ovarian GCs
To determine the effect of RSPO2 on the apoptosis of GCs, the apoptosis rate and expression of genes related to apoptosis pathways were detected after overexpression and knockdown of RSPO2. RSPO2 overexpression significantly inhibited the mRNA and protein expression level of pro-apoptosis-related genes (Caspase3 and BAX; Fig. 3A, B) and significantly promoted the protein expression levels of anti-apoptosis-related gene (BCL2; Fig. 3B). To further confirm the effects of RSPO2 on apoptosis, the cleaved protein expression of Caspase3, Caspase8, Caspase9, and PARP in GCs were detected. Results showed that RSPO2 overexpression significantly inhibited the protein expression of cleaved Caspase3, cleaved Caspase8, cleaved Caspase9, and cleaved PARP (Fig. 3C) and the apoptosis of GCs (Fig. 3D). Moreover, RSPO2 silencing significantly promoted the mRNA expression level of BAX (Fig. 3E) as well as the protein expression level of Caspase3, BAX, cleaved Caspase3, cleaved Caspase9, and cleaved PARP (Fig. 3F, G) and significantly promoted the apoptosis of GCs (Fig. 3H). Similarly, RSPO2-siRNA5 also significantly promoted the mRNA expression of Caspase3 and BAX (Fig. 3I) as well as the apoptosis of GCs (Fig. 3J). Collectively, these results suggested that RSPO2 inhibited the apoptosis of porcine ovarian GCs.

A–C The mRNA (A) and protein (B) expression levels of Caspase3, Caspase8, Caspase9, BAX, and BCL2, as well as the cleaved protein (C) expression levels of Caspase3, Caspase8, Caspase9, and PARP in porcine GCs with RSPO2 overexpression. D The apoptosis rates of GCs with RSPO2 overexpression was assessed by flow cytometry. E–G The mRNA (E) and protein (F) expression levels of Caspase3, Caspase8, Caspase9, BAX, and BCL2, as well as the cleaved protein (G) expression levels of Caspase3, Caspase8, Caspase9, and PARP in porcine GCs with RSPO2 knockdown. H The apoptosis rates of GCs with RSPO2 knockdown was assessed by flow cytometry. I The mRNA expression levels of Caspase3, Caspase8, Caspase9, BAX, and BCL2 in porcine GCs transfected with RSPO2-siRNA5. J The apoptosis rates of GCs transfected with RSPO2-siRNA5 was assessed by flow cytometry. *P < 0.05, **P < 0.01.
RSPO2 promoted the secretion of E2 in porcine ovarian GCs
To determine the effect of RSPO2 on the E2 secretion of GCs, the expression of genes related to estrogen signaling pathway were detected after overexpression and knockdown of RSPO2. qRT-PCR (Fig. 4A) and WB (Fig. 4B) results indicated that RSPO2 overexpression significantly promoted the mRNA and protein levels expression of CYP19A1 and HSD17B1, while RSPO2 silencing significantly inhibited the mRNA and protein expression levels of HSD17B1 as well as the protein expression level of CYP19A1. Moreover, RSPO2 overexpression significantly promoted the secretion of E2 in GCs, while RSPO2 knockdown displayed the opposite effects (Fig. 4C). Similarly, RSPO2-siRNA5 also significantly inhibited the mRNA expression of CYP19A1 and HSD17B1 (Fig. 4D) and the secretion of E2 in GCs. (Fig. 4E). In summary, RSPO2 promoted the secretion of E2 in porcine ovarian GCs.

A, B The mRNA (A) and protein (B) expression levels of CYP19A1, STAR, HSD17B1, and HSD3B1 in porcine GCs with RSPO2 overexpression and inhibition. C The concentration of E2 in porcine ovarian GCs with RSPO2 overexpression and inhibition were assessed by ELISA. D The mRNA expression levels of CYP19A1, STAR, HSD17B1, and HSD3B1 in porcine GCs transfected with RSPO2-siRNA5. E The concentration of E2 in porcine ovarian GCs transfected with RSPO2-siRNA5 was assessed by ELISA. *P < 0.05, **P < 0.01.
Inhibition of RSPO2 blocked the development of porcine follicles
To further investigate the biological function of RSPO2 in the development of follicles, the lentiviral vector knockdown of RSPO2 (sh-RSPO2) and negative control (sh-NC) were built and transfected into porcine follicles cultured in vitro. The mRNA and protein expression levels of RSPO2 in sh-RSPO2 group were significantly lower than those in the sh-NC group (Fig. 5A, B), indicating that sh-RSPO2 has been successfully transfected into follicles. Moreover, RSPO2 knockdown decreased the mRNA and protein expression levels of PCNA, CCND1, CYP19A1, LGR4, and CTNNB1, while increased the mRNA and protein expression levels of Caspase3 in porcine follicles (Fig. 5A, B). Besides, RSPO2 knockdown promoted the loss of follicular blood vessels and the opacity of follicular fluid (Fig. 5C), and terminal deoxynucleotidyl transferase-mediated dUTP-fluorescein nick end labeling (TUNEL) assay showed that the apoptosis of GCs was markedly increased (Fig. 5D). These results indicated that inhibition of RSPO2 might block the development of porcine follicles by promoting the apoptosis of GCs.

A, B The mRNA (A) and protein (B) expression levels of RSPO2, Caspase3, PCNA, CCND1, CYP19A1, LGR4, and CTNNB1 in porcine follicle with RSPO2 inhibition. The numbers below bands were folds of band intensities relative to control. The band intensities were measured by ImageJ and normalized to Tubulin. C The photos of porcine follicles cultured in vitro after transfected with sh-RSPO2 on first, fourth, and seventh day. D The apoptosis of follicular GCs was detected by TUNEL after transfected with sh-RSPO2 at the fourth day. *P < 0.05, **P < 0.01.
Silencing RSPO2 blocked follicular development and onset of puberty in mice
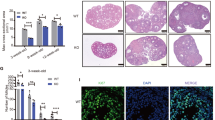
To further verify the role of RSPO2 on follicular development and the onset of puberty, the sh-RSPO2 lentiviral vector was injected into mice via intraperitoneal injection. Then the mice were slaughtered, and we found that the mRNA and protein expression levels of RSPO2 in the ovaries of mice transfected with sh-RSPO2 were significantly lower than those mice transfected with sh-NC (Fig. 6A, B), indicating that sh-RSPO2 had been successfully transfected into mice. Meanwhile, RSPO2 knockdown decreased the mRNA and protein expression levels of CCND1, CYP19A1, LGR4, and CTNNB1, while increased the mRNA expression level of Caspase3 as well as protein expression level of cleaved Caspase3 in the mouse ovaries (Fig. 6A, B). Next, the age of pubertal initiation and serum E2 level of mice transfected with sh-RSPO2 were analyzed, the development status of ovarian follicles was detected by hematoxylin and eosin staining, and the apoptosis of GCs in follicles was examined by TUNEL. The mice injected with sh-RSPO2 had delayed vaginal opening (Fig. 6C; mean age at vaginal opening: blank, 38 ± 2.12 days; sh-NC, 38.8 ± 1.48 days; sh-RSPO2, 46.8 ± 2.68 days; P < 0.01). RSPO2 knockdown significantly decreased the serum E2 concentration in mice (Fig. 6D). Examination of the ovaries at age 50 days showed that sh-RSPO2-treated mice had numerous antral follicles but few corpora lutea, indicating that they had not ovulated and consequently that pubertal initiation failed (Fig. 6E). In contrast, the ovaries of sh-NC-treated and untreated mice exhibited many corpora lutea but few antral follicles, indicating that they ovulated and the pubertal initiation had occurred (Fig. 6E). Moreover, RSPO2 knockdown increased the apoptosis of ovarian GCs in mice (Fig. 6F). These results indicated that silencing RSPO2 might inhibit the development of follicles and eventually delay the onset of puberty by promoting the apoptosis of GCs and inhibiting the secretion of E2 in mice.

A, B The mRNA (A) and protein (B) expression levels of RSPO2, Caspase3, cleaved Caspase3, CCND1, CYP19A1, LGR4, and CTNNB1 in the ovaries of mice with RSPO2 inhibition. The numbers below bands were folds of band intensities relative to control. The band intensities were measured by ImageJ and normalized to Tubulin. C The percentage of mice at vaginal opening in the blank, sh-NC, and sh-RSPO2 groups (n = 5). D The E2 concentration in serum from blank, sh-NC and sh-RSPO2-injected mice collected at age 50 days was detected by ELISA. E Example of the ovaries from blank, sh-NC, and sh-RSPO2-injected mice collected at age 50 days. CL indicates corpora lutea and arrows point to example of antral follicles. F The apoptosis of follicular GCs from blank, sh-NC, and sh-RSPO2-injected mice collected at age 50 days was detected by TUNEL. *P < 0.05, **P < 0.01.
Hypomethylation of CGI2 recruited E2F1 to promote the transcription of RSPO2
To explore the mechanism of DNA methylation that regulated RSPO2 expression, PROMO website was used to predict the potential transcription factor-binding sites of the CGI region. There were one potential binding site of E2F1 on the CGI1 region (Site1, −1239/−1232 bp, Fig. 7A) and two potential binding sites of E2F1 on the CGI2 region (Site2, −758/−749 bp and Site3, −563/−553 bp, Fig. 7A). Then the expression of RSPO2 in GCs treated with DNMT1-siRNA was detected, and DNMT1 knockdown significantly increased the mRNA and protein levels of RSPO2 (Fig. 7B). Bisulfite sequencing PCR (BSP) was used to measure the methylation level changes of NCGI1, NCGI2, NCGI3, CGI1, CGI2-1, and CGI2-2 after DNMT1. As shown in Fig. 7C, the methylation levels of Site2 and Site3 were obviously reduced, but there was no difference at Site1 (Fig. 7C). Chromatin immunoprecipitation (ChIP) and ChIP-qPCR were further performed to detect whether the occupancy of E2F1 to CGI was regulated by DNA methylation. Results showed that the enrichment and occupancy of E2F1 at Site2 and Site3 were significantly increased after DNMT1 knockdown, but there was no difference at Site1 (Fig. 7D, E). These observations suggested that DNA methylation might regulate RSPO2 expression by mediating the occupancy of E2F1 to CGI2 at Site2 and Site3.

A Predicted E2F1-binding sites in the CGI1 and CGI2 region of RSPO2 promoter. B The mRNA and protein expression level of RSPO2 in porcine GCs treated with DNMT1-siRNA. The numbers below bands were folds of band intensities relative to control. The band intensities were measured by ImageJ and normalized to Tubulin. C BSP was used to detect the NCGI and CGI methylation status of RSPO2 in porcine GCs treated with DNMT1-siRNA. The red box showed the three potential binding sites region of E2F1 in the RSPO2 promoter CGI. D, E The enrichment of E2F1 on the CGI region after DNMT1 knockdown was detected by ChIP (D) and ChIP-qPCR (E). F The mRNA and protein expression levels of E2F1 after transfection with pcDNA3.1 or pcDNA3.1-E2F1 in porcine GCs. G The luciferase reporter plasmids carrying CGI2 region of RSPO2 were methylated in vitro and co-transfected with pcDNA3.1-E2F1 into GCs. Relative luciferase activity in GCs was determined. **P < 0.01.
Moreover, the overexpression vector of E2F1 (pcDNA3.1-E2F1) were constructed, and the effect of E2F1 on the transcriptional activity of RSPO2 with methylated or unmethylated CGI2 were detected by dual-luciferase activity analysis. E2F1 overexpression significantly increased the mRNA and protein levels of E2F1 in GCs (Fig. 7F). Furthermore, E2F1 overexpression significantly increased the transcription activity of the unmethylated pGL3-CGI2-1 by 6.6 times, while only 1.8 times in the methylated pGL3-CGI2-1. Similarly, E2F1 overexpression significantly increased the transcription activity of the unmethylated pGL3-CGI2–2 by 12.8 times, while only 1.9 times in the methylated pGL3-CGI2–2 (Fig. 7G). These results implied that the hypomethylation of CGI2 recruited E2F1 to promote the transcription of RSPO2.
Discussion
Accumulating studies have shown that the survival of GCs determines the fate of follicles [34, 35]. The continuous proliferation and E2 secretion of GCs promotes the growth of follicles [36, 37], while the excessive apoptosis of GCs causes follicular atresia [38], retards the development of follicles, and even leads to the delay of sexual maturity [39] and reduction of reproductive performance in mammals [40]. The transcription factor E2F1, a member of E2F family, has been highly suggested to be involved in the ovarian follicle development by regulating the proliferation and E2 secretion of GCs in cattle [41]. Previous studies have recommended that the RSPO2 gene regulates the GC proliferation and promotes follicular development in humans [32] and mice [33]. Altering the DNA methylation status of GCs is involved in ovarian follicle development by regulating the expression level of genes, e.g., AKR1C3, HAPLN1, and PTGER1 [27]. In this study, we found that the hypomethylation of −758/−749 and −563/−553 regions in RSPO2 promoter facilitated the binding of E2F1 and promoted the transcription of RSPO2, which further promoted the proliferation and steroid hormone secretion of GCs, inhibited the apoptosis of GCs, and ultimately promoted follicular development and pubertal initiation (Fig. 8).

The hypomethylation of CGI2 in RSPO2 promoter facilitated the occupancy of E2F1 and enhanced the transcription of RSPO2, which further promoted the proliferation and E2 secretion of GCs, inhibited the apoptosis of GCs and ultimately ameliorated the development of follicles and pubertal initiation through Wnt signaling pathway.
DNA methylation is a widely studied epigenetic phenomenon, usually associated with gene silencing [42]. 5-Aza-CdR reduced the hypermethylation level of RSPO2 and induced its re-expression in gastric cancer cell lines [43]. In this study, we found that the expression level of RSPO2 was negatively correlated with the methylation level of the promoter CGI during follicular development. (Fig. 1B, C). Moreover, both inhibition of DNA methylation (Fig. 1E) and DNMT1 knockdown (Fig. 7C) reduced the methylation levels of RSPO2 promoter and promoted the mRNA and protein expression levels of RSPO2 in porcine GCs (Figs. 1D and 7B). Thus, we hypothesized that DNA methylation might regulate RSPO2 expression level in porcine GCs. To further explore the core regions of DNA methylation regulating RSPO2 expression level, the RSPO2 promoter was segmented (Fig. 1A). The dual-luciferase activity results showed that the DNA hypermethylation of the CGI2 region induced a prominent lowering of RSPO2 promoter activity (Fig. 1F). These observations indicated that DNA methylation status of CGI2 region might regulate RSPO2 expression level in porcine GCs.
Moreover, we explored how DNA methylation regulated the expression level of RSPO2. Numerous studies have shown that the hypermethylated state of CG dinucleotide may regulate gene expression by hindering the binding of transcription factors [44,45,46]. In temozolomide-sensitive glioblastoma cell, the CGI hypermethylation of SNHG12 promoter blocked the occupancy of SP1 and inhibited the expression level of SNHG12 [44]. Thus, we predicted the potential transcription factor-binding site on all CG dinucleotide within the CGI region of RSPO2 promoter and found that transcription factor E2F1 had the most binding sites in this region (Fig. 7A). Moreover, DNMT1 knockdown reduced the methylation level of E2F1-binding sites at CGI2 (Fig. 7C) and significantly increased the expression of RSPO2 (Fig. 7B). That DNMT1 knockdown promoted the binding of E2F1 to the RSPO2 promoter CGI2 region was further determined by ChIP (Fig. 7D) and ChIP-qPCR (Fig. 7E). Further, E2F1 overexpression significantly promoted the activity of unmethylated CGI2 region in RSPO2 promoter compared to methylated CGI2 region in porcine GCs (Fig. 7G). These observations indicated that DNA methylation might partially inhibit the expression level of RSPO2 by hindering E2F1 binding to the CGI2 region of RSPO2 promoter.
It was widely known that abnormal proliferation and apoptosis can affect E2 secretion of GCs and follicular development. Our data showed that RSPO2 promoted the proliferation of GCs, inhibited the apoptosis of GCs, and promoted the secretion of E2. PCNA [47], CDK1 [48], and CCND1 [49], all known to be proliferation regulators, have been confirmed to promote GC proliferation and follicular development in mammals. The aromatase encoded by CYP19A1 is a key rate-limiting enzyme for E2 biosynthesis [50, 51]. Upregulation of CYP19A1 expression stimulates E2 release and inhibits porcine GC apoptosis and follicular atresia [52]. Activation of BAX stimulates Caspase3/9 and then promotes the cleavage of PARP, thereby inducing the apoptotic process [53]. The decreased expression of BAX inhibited GC apoptosis in rat [54], and knockout of BAX in mice resulted in an increase in ovarian follicle numbers [55]. Decreased expression of Caspase3 inhibited apoptosis of premature ovarian insufficiency rat ovaries to restore ovarian function and structure [56]. Further, we detected the expression level of several key genes in the signaling pathways associated with proliferation, apoptosis, and steroid hormone secretion after overexpression or knockdown of RSPO2 in GCs. Among these genes, the expression levels of proliferation markers PCNA, CDK1, and CCND1 were significantly increased by pcDNA3.1-RSPO2 (Fig. 2C, D), and these were significantly decreased by RSPO2-siRNA (Fig. 2G, H). The expression levels of pro-apoptotic genes Caspase3, cleaved Caspase3, cleaved Caspase9, cleaved PARP, and BAX were significantly decreased by pcDNA3.1-RSPO2 (Fig. 3B, C), and these were significantly increased by RSPO2-siRNA (Fig. 3E, G). The mRNA and protein levels of steroid hormone secretion markers CYP19A1 and HSD17B1 were significantly increased by pcDNA3.1-RSPO2, and these were significantly decreased by RSPO2-siRNA (Fig. 4A, B). Overall, RSPO2 might promote the proliferation and inhibit the apoptosis and secretion of E2 in porcine GCs.
Previous studies have confirmed that healthy follicles were characterized with a well-vascularized follicular wall and the clarity of follicular fluid, and atretic follicles were characterized with no blood vessels and the opacification of follicular fluid [57, 58]. The apoptosis of GCs is the main cause of follicular atresia and selection [59, 60]. In this study, we found that RSPO2 knockdown promoted apoptosis of porcine follicular GCs (Fig. 5D), the loss of follicular blood vessels, and the opacification of follicular fluid (Fig. 5C). Similarly, RSPO2 knockdown reduced the E2 concentration in serum (Fig. 6D), blocked most of the follicles in the antral follicle stage (Fig. 6E), and promoted the apoptosis of follicular GCs in mice (Fig. 6F). Additionally, studies have indicated that RSPO2-LGRs could activate and enhance the Wnt/β-catenin signaling pathway [61] to participate in GC apoptosis [62], proliferation, and follicle development [33]. In this study, we found that inhibition of RSPO2 decreased the mRNA and protein expression levels of LGR4 and CTNNB1 in the porcine follicles (Fig. 5A, B) and mice ovaries (Fig. 6A, B). Interestingly, we also found that RSPO2 knockdown delayed the onset of mouse puberty (Fig. 6C). Therefore, we speculate that inhibition of RSPO2 may delay the initiation of puberty by inhibiting follicle development. In summary, inhibition of RSPO2 might promote the apoptosis of follicular GCs through the Wnt signaling pathway, thereby inhibiting the development of follicles in mammals.
Materials and methods
Cell and tissue culture
The ovaries of pre-puberty sows were collected from a local slaughterhouse and transported to the laboratory using phosphate-buffered saline (PBS) containing penicillin (100 IU/mL) and streptomycin (100 μg/mL) (Invitrogen, Shanghai, China). Subsequently, the GCs were aspirated by inserting a syringe into a 3–5-mm follicle and washed twice with PBS. The cells were then seeded into culture flasks containing 10% fetal bovine serum (Hyclone, Logan, UT, USA) in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, Logan, UT, USA) and 100 IU/mL penicillin and 100 μg/mL streptomycin and finally incubated at 37 °C under 5% CO2.
Antral follicles 3–5 mm in diameter were removed from the ovaries using forceps and scalpels. Then the follicles were cleaned with PBS and follicular culture medium, respectively. Follicular culture medium was serum-free DMEM/F12. Twenty-four-well plates were used for culture, with one follicle per well, and incubated at 38.5 °C under 5% CO2.
Quantitative reverse transcription PCR
TRIzol reagent (TaKaRa, Tokyo, Japan) was used to extract total RNA from the sample, and then the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA) was used to reverse-transcribe the mRNAs. Maxima SYBR Green qRT-PCR Master Mix (2×) (Thermo Scientific, Waltham, MA, USA) was used to quantify the relative expression levels of mRNAs in the CFX96 Touch Real-Time PCR system (Bio-Rad, Berkeley, CA, USA). Using the expression level of glyceraldehyde phosphate dehydrogenase as endogenous control, the relative expression level of genes was calculated with the 2−ΔΔct method. The primer sequences used in pig are listed in Table 1 and that used in mouse are listed in Table 2.
EdU and MTT assay
The Cell-LightTM Edu Apollo 567 In Vitro Kit (RiboBio, Guangdong, China) was used to analyze cell proliferation. Briefly, GCs were cultured in 48-well plates and transfected with plasmid for 24 h. GCs were incubated at room temperature with 50 μM EDU for 2 h, washed twice with PBS, and then incubated with 80% acetone for 30 min. After GCs were washed twice by PBS, 0.5% Triton X-100 was added for 10 min, 1× Apollo was incubated in darkness for 30 min, and Hoechst was incubated for 30 min. Finally, three fields were randomly selected from each well and GCs were counted under an inverted fluorescence microscope.
The MTT Cell Proliferation and Cytotoxicity Assay Kit (Beyotime, Shanghai, China) was used to analyze cell proliferation. Briefly, GCs were cultured in 10 μL (5 mg/mL) MTT solution for 4 h and then added 100 μL Formazan solution for 4 h in 96-well plates. Finally, the absorbance of 570 nm was measured.
Flow cytometry
The Annexin V-FITC Apoptosis Detection Kit (BioVision, Milpitas, CA, USA) was used to analyze cell apoptosis. Briefly, GCs were cultured in 6-well plates and transfected with plasmids for 24 h. The collected cells were centrifuged at 1000 rpm for 5 min, the supernatant was discarded, and washed twice with PBS. Then, 500 μL of 1× Annexin V buffer was added to gently resuspend the cells, and 5 μL of Annexin V–fluorescein isothiocyanate and 5 μL of propidium iodide (PI) staining solution were added and mixed. Finally, flow cytometry was performed after incubation for 15 min at room temperature in darkness. For results, the figure has four quadrants, the lower right quadrant is annexin-positive/PI-negative early apoptotic cells, the upper right quadrant is annexin-positive/PI-positive late apoptotic cells, the lower left quadrant is living cell, and the upper left quadrant is mechanical injury cells. In this study, the apoptotic ratio of GCs is the sum of early and late apoptosis.
Enzyme-linked immunosorbent assay (ELISA)
The concentration of E2 in the serum of mice and supernatant of cell culture medium was detected using a mouse or porcine E2 ELISA Kit (Jingmei Biotechnology, Jiangshu, China) performed according to the manufacturer’s instructions. Fifty microliters of standard with different concentrations were added into the standard well, and 50 μL of samples to be tested were added into the sample well. Then 100 μL of horseradish peroxidase (HRP) was added to each well and incubated for 60 min at 37 °C. After cleaning, 50 μL of substrate A and B were added and incubated for 15 min at 37 °C. Finally, termination solution was added to measure the optical density value at 450 nm.
Plasmid construction and dual-luciferase reporter assay
The promoter (−2232/+66 bp), non-CpG island 1 (NCGI1) (−1540/−1357 bp), CGI1 (−1345/−1165 bp), NCGI2 (−1187/−1003 bp), CGI2-1 (−1014/−721 bp), CGI2–2 (−709/−362 bp), and NCGI3 (−327/−76 bp) regions of RSPO2 were amplified from genomic DNA of GCs, followed by cloning into the pGL3 vector to obtain pGL3-RSPO2, pGL3-NCGI1, pGL3-CGI1, pGL3-NCGI2, pGL3-CGI2-1, pGL3-CGI2–2, and pGL3-NCGI3 recombinant vectors. The pGL3-basic and pGL3-control were used as the control group. Then the CpG transmethylase M.SssI (M0226, NEW ENGLAND Biolabs, Beijing, China) was incubated with these 9 vectors for 1 h at 37 °C to methylate all CpG residues and the corresponding unmethylated group was incubated with nuclease-free water. The BstU I endonuclease was incubated with these vectors for 1 h at 60 °C to detect the methylation status.
The CDS region of RSPO2 and E2F1 gene were amplified from cDNA of GCs and cloned into the expression vector pcDNA3.1 (Invitrogen). These vectors were purified by the HiPure Plasmid EF Micro Kit (P1111-03, Magen, Guangzhou, China). All siRNAs were designed and synthesized by RiboBio (Guangzhou, China). Plasmids were transfected into 80% confluent GCs for 24 h using LipofectamineTM 3000 Reagent (Thermo Scientific, Waltham, MA, USA). All luciferase activities were measured with the BioTek Synergy 2 multifunctional microplate reader (BioTek, Winooski, VT, USA) by using a Dual-luciferase Reporter Assay Kit (Promega, Madison, WI, USA) and normalized to Renilla luciferase activity. The primer sequences are listed in Table 3.
Animals and lentivirus delivery
Four-week-old female C57BL/6J mice (n = 15) were purchased from Guangdong Medical Laboratory Animal Center (Guangzhou, China) and were randomly divided into blank (n = 5), sh-NC (n = 5), and sh-RSPO2 (n = 5) groups with 5 in each group.
The CDS region or siRNA of RSPO2 was inserted into a GV208 lentivirus vector containing a CMV-driven EGFP reporter gene and a Ubi promoter upstream of restriction sites (Age I and BamH I). All constructs were verified by sequencing. Then recombinant lentiviruses were produced by co-transfecting 293T cells. GFP expression was measured in 293T cells to determine the virus titers, expressed as transducing units (TU) per mL. The titers of lentiviral vector that overexpress or interfere with RSPO2 (LV-RSPO2 or sh-RSPO2) was approximately 1E + 9 TU/ml. In all, 1 × 107 TU of lentivirus was then injected into the mice of each group (n = 5) through intraperitoneal injection. The injection was given once a week for 3 weeks.
Bisulfite sequencing PCR
Genomic DNA was extracted from GCs and follicles with a Tissue DNA Kit (D3396-02, Omega Bio-tek, USA). The purified DNA was exposed to bisulfite with an EZ DNA Methylation-GoldTM Kit (D5006, ZYMO RESEARCH, CA, USA) according to the manufacturer’s protocol. Then the corresponding fragment was amplified by BSP primer using the bisulfite-converted DNA as template. Finally, the sequencing results are compared with the original sequence through the QUMA website (http://quma.cdb.riken.jp/) and plotted. Ten clones were needed for each sample to calculate the methylation rate. The specific primers used for BSP are listed in Table 4.
TUNEL assay
Apoptotic cells were detected by the TUNEL assays using a One Step TUNEL Apoptosis Assay Kit (Beyotime Biotech, Jiangshu, China) according to the manufacturer’s protocol. Briefly, the different treated follicles were made into paraffin sections and then treated with xylene, ethanol, and protease K, respectively. After being washed by PBS, the sections were treated with the freshly prepared TUNEL reaction mixture. Images were obtained by using a Nikon ECLIPSE Ti2 fluorescence microscope.
ChIP assay
ChIP assay was conducted using the PierceTM ChIP Kit (ThermoFisher, Rockford, IL, USA) according to the manufacturer’s instructions. The cultured GCs were incubated with 1% formaldehyde at room temperature for 10 min. After GCs were quenched with glycine, the chromatin fragments of GCs were extracted with automatic ultrasonic crushing apparatus. The specific E2F1 antibody (ab179445, Abcam, Cambridge, MA, USA) and IgG antibody (12–370, Millipore) were then incubated with the cell fragmentation solution at 4 °C overnight. The immunoprecipitated DNA was purified for PCR and qPCR analyses, and the specific primers are listed in Table 5.
WB analysis
The total protein was extracted and the concentration was detected by a BCA Protein Assay Kit (Vigorous Bio-technology Beijing Co., Ltd., Beijing, China). Equal amounts of sample protein were electrophoresed on 4–20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride (PVDF) membranes. Then the PVDF membranes were sealed with Tris-buffered saline-Tween (TBST) solution, which contained 5% skimmed milk at room temperature for 1–2 h. After the PVDF membranes were washed with TBST for 3 times, the membranes were incubated overnight at 4 °C with the following diluted primary antibody: RSPO2 (17781-1-AP, Proteintech, 1:1000), E2F1 (ab179445, Abcam, 1:2000), PCNA (10205-2-AP, Proteintech, 1:2000), CDK1 (19532-1-AP, Proteintech, 1:1000), CCND1 (26939-1-AP, Proteintech, 1:1000), Caspase3 (19677-1-AP, Proteintech, 1:1000), cleaved Caspase3 (#9661, CST, 1:1000), cleaved Caspase8 (#8592, CST, 1:1000), cleaved Caspase9 (#9509, CST, 1:1000), cleaved PARP (#5625, CST, 1:1000), BAX (50599-2-Ig, Proteintech, 1:5000), BCL2 (12789-1-AP, Proteintech, 1:2000), CYP19A1 (bs-0114R, Bioss, 1:1000), HSD17B1 (bs-6603R, Bioss, 1:1000), LGR4 (20150-1-AP, Proteintech, 1:1000), CTNNB1 (51067-2-AP, Proteintech, 1:10,000), and Tubulin (11224-1-AP, Proteintech, 1:5000). The membranes were washed with TBST for 3 times and incubated with goat anti-rabbit IgG H&L (HRP) (ab205718, Abcam, 1:10,000) or goat anti-mouse IgG H&L (HRP) (ab6789, Abcam, 1:5000) for 2 h at room temperature. The blots were visualized by the Odyssey Fc Imaging System (LI-COR Biosciences, Lincoln, NE), and the intensity of protein bands was measured by the NIH ImageJ software.
Statistical analysis
In all the panels, data are shown as mean ± standard deviation from at least three biological replicates. Statistical significance of differences between means was analyzed by Student’s t test (**P < 0.05 and *P < 0.01).
References
Oktem O, Urman B. Understanding follicle growth in vivo. Hum Reprod. 2010;25:2944–54.
Yuan X, Ye S, Chen Z, Pan X, Huang S, Li Z, et al. Dynamic DNA methylation of ovaries during pubertal transition in gilts. BMC Genomics. 2019;20:510.
Lapatto R, Pallais JC, Zhang D, Chan YM, Mahan A, Cerrato F, et al. Kiss1-/- mice exhibit more variable hypogonadism than Gpr54-/- mice. Endocrinology. 2007;148:4927–36.
Hsueh AJ, Kawamura K, Cheng Y, Fauser BC. Intraovarian control of early folliculogenesis. Endocr Rev. 2015;36:1–24.
Hu J, Jin J, Qu Y, Liu W, Ma Z, Zhang J, et al. ERO1alpha inhibits cell apoptosis and regulates steroidogenesis in mouse granulosa cells. Mol Cell Endocrinol. 2020;511:110842.
Kranc W, Budna J, Kahan R, Chachula A, Bryja A, Ciesiolka S, et al. Molecular basis of growth, proliferation, and differentiation of mammalian follicular granulosa cells. J Biol Regul Homeost Agents. 2017;31:1–8.
Kim JM, Boone DL, Auyeung A, Tsang BK. Granulosa cell apoptosis induced at the penultimate stage of follicular development is associated with increased levels of Fas and Fas ligand in the rat ovary. Biol Reprod. 1998;58:1170–6.
Yeung CK, Wang G, Yao Y, Liang J, Tenny Chung CY, Chuai M, et al. BRE modulates granulosa cell death to affect ovarian follicle development and atresia in the mouse. Cell Death Dis. 2017;8:e2697.
Emmen JM, Couse JF, Elmore SA, Yates MM, Kissling GE, Korach KS. In vitro growth and ovulation of follicles from ovaries of estrogen receptor (ER){alpha} and ER{beta} null mice indicate a role for ER{beta} in follicular maturation. Endocrinology. 2005;146:2817–26.
Couse JF, Yates MM, Deroo BJ, Korach KS. Estrogen receptor-beta is critical to granulosa cell differentiation and the ovulatory response to gonadotropins. Endocrinology. 2005;146:3247–62.
Hegele-Hartung C, Siebel P, Peters O, Kosemund D, Muller G, Hillisch A, et al. Impact of isotype-selective estrogen receptor agonists on ovarian function. Proc Natl Acad Sci USA. 2004;101:5129–34.
Khristi V, Chakravarthi VP, Singh P, Ghosh S, Pramanik A, Ratri A, et al. ESR2 regulates granulosa cell genes essential for follicle maturation and ovulation. Mol Cell Endocrinol. 2018;474:214–26.
Maneix L, Antonson P, Humire P, Rochel-Maia S, Castaneda J, Omoto Y, et al. Estrogen receptor beta exon 3-deleted mouse: the importance of non-ERE pathways in ERbeta signaling. Proc Natl Acad Sci USA. 2015;112:5135–40.
Zhang H, Luo Q, Lu X, Yin N, Zhou D, Zhang L, et al. Effects of hPMSCs on granulosa cell apoptosis and AMH expression and their role in the restoration of ovary function in premature ovarian failure mice. Stem Cell Res Ther. 2018;9:20.
Pan Z, Zhang J, Li Q, Li Y, Shi F, Xie Z, et al. Current advances in epigenetic modification and alteration during mammalian ovarian folliculogenesis. J Genet Genomics. 2012;39:111–23.
LaVoie HA. Epigenetic control of ovarian function: the emerging role of histone modifications. Mol Cell Endocrinol. 2005;243:12–8.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Lomniczi A, Loche A, Castellano JM, Ronnekleiv OK, Bosch M, Kaidar G, et al. Epigenetic control of female puberty. Nat Neurosci. 2013;16:281–9.
Lomniczi A, Wright H, Ojeda SR. Epigenetic regulation of female puberty. Front Neuroendocrinol. 2015;36:90–107.
Yang C, Gao X, Ye J, Ding J, Liu Y, Liu H, et al. The interaction between DNA methylation and long non-coding RNA during the onset of puberty in goats. Reprod Domest Anim. 2018;53:1287–97.
Li J, Bian EB, He XJ, Ma CC, Zong G, Wang HL, et al. Epigenetic repression of long non-coding RNA MEG3 mediated by DNMT1 represses the p53 pathway in gliomas. Int J Oncol. 2016;48:723–33.
Geng X, Zhao J, Huang J, Li S, Chu W, Wang WS, et al. lnc-MAP3K13-7:1 inhibits ovarian GC proliferation in PCOS via DNMT1 downregulation-mediated CDKN1A promoter hypomethylation. Mol Ther. 2020;29:1279–93.
Zhang Y, Zhang S, Liu Z, Zhang L, Zhang W. Epigenetic modifications during sex change repress gonadotropin stimulation of cyp19a1a in a teleost ricefield eel (Monopterus albus). Endocrinology. 2013;154:2881–90.
Vanselow J, Spitschak M, Nimz M, Furbass R. DNA methylation is not involved in preovulatory down-regulation of CYP11A1, HSD3B1, and CYP19A1 in bovine follicles but may have a role in permanent silencing of CYP19A1 in large granulosa lutein cells. Biol Reprod. 2010;82:289–98.
Guo Y, Wang W, Chen Y, Sun Y, Li Y, Guan F, et al. Continuous gibberellin A3 exposure from weaning to sexual maturity induces ovarian granulosa cell apoptosis by activating Fas-mediated death receptor signaling pathways and changing methylation patterns on caspase-3 gene promoters. Toxicol Lett. 2020;319:175–86.
Sagvekar P, Mangoli V, Desai S, Patil A, Mukherjee S. LINE1 CpG-DNA hypomethylation in granulosa cells and blood leukocytes is associated with PCOS and related traits. J Clin Endocrinol Metab. 2017;102:1396–405.
Sagvekar P, Kumar P, Mangoli V, Desai S, Mukherjee S. DNA methylome profiling of granulosa cells reveals altered methylation in genes regulating vital ovarian functions in polycystic ovary syndrome. Clin Epigenet. 2019;11:61.
Carmon KS, Gong X, Lin Q, Thomas A, Liu Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc Natl Acad Sci USA. 2011;108:11452–7.
Glinka A, Dolde C, Kirsch N, Huang YL, Kazanskaya O, Ingelfinger D, et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 2011;12:1055–61.
de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014;28:305–16.
Liu Q, Zhao Y, Xing H, Li L, Li RX, Dai J, et al. The role of R-spondin 1 through activating Wnt/beta-catenin in the growth, survival and migration of ovarian cancer cells. Gene. 2019;689:124–30.
Cheng Y, Kawamura K, Takae S, Deguchi M, Yang Q, Kuo C, et al. Oocyte-derived R-spondin2 promotes ovarian follicle development. FASEB J. 2013;27:2175–84.
De Cian MC, Gregoire EP, Le Rolle M, Lachambre S, Mondin M, Bell S, et al. R-spondin2 signaling is required for oocyte-driven intercellular communication and follicular growth. Cell Death Differ. 2020;27:2856–71.
Quirk SM, Cowan RG, Harman RM, Hu CL, Porter DA. Ovarian follicular growth and atresia: the relationship between cell proliferation and survival. J Anim Sci. 2004;82:E40–52.
Matsuda F, Inoue N, Manabe N, Ohkura S. Follicular growth and atresia in mammalian ovaries: regulation by survival and death of granulosa cells. J Reprod Dev. 2012;58:44–50.
Hillier SG. Gonadotropic control of ovarian follicular growth and development. Mol Cell Endocrinol.2001;179:39–46.
Jiang JY, Cheung CK, Wang Y, Tsang BK. Regulation of cell death and cell survival gene expression during ovarian follicular development and atresia. Front Biosci. 2003;8:d222–37.
Regan SLP, Knight PG, Yovich JL, Leung Y, Arfuso F, Dharmarajan A. Granulosa cell apoptosis in the ovarian follicle-a changing view. Front Endocrinol. 2018;9:61.
Phoophitphong D, Srisuwatanasagul S, Koonjaenak S, Tummaruk P. Apoptotic cell localization in preantral and antral follicles in relation to non-cyclic and cyclic gilts. Reprod Domest Anim. 2016;51:400–6.
Drummond AE. The role of steroids in follicular growth. Reprod Biol Endocrinol. 2006;4:16.
Morrell BC, Perego MC, Maylem ERS, Zhang L, Schutz LF, Spicer LJ. Regulation of the transcription factor E2F1 mRNA in ovarian granulosa cells of cattle. J Anim Sci. 2020;98:skz376.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38.
Wilhelm F, Simon E, Boger C, Behrens HM, Kruger S, Rocken C. Novel insights into gastric cancer: methylation of r-spondins and regulation of LGR5 by SP1. Mol Cancer Res. 2017;15:776–85.
Lu C, Wei Y, Wang X, Zhang Z, Yin J, Li W, et al. DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol Cancer. 2020;19:28.
Chen PY, Feng S, Joo JW, Jacobsen SE, Pellegrini M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011;12:R62.
Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551–65.
Oktay K, Schenken RS, Nelson JF. Proliferating cell nuclear antigen marks the initiation of follicular growth in the rat. Biol Reprod. 1995;53:295–301.
Chen S, Wang F, Liu Z, Zhao Y, Jiang Y, Chen L, et al. Brain-derived neurotrophic factor promotes proliferation and progesterone synthesis in bovine granulosa cells. J Cell Physiol. 2019;234:8776–87.
Liu C, Pan B, Yang L, Wang B, Li J. Beta defensin 3 enhances ovarian granulosa cell proliferation and migration via ERK1/2 pathway in vitrodagger. Biol Reprod. 2019;100:1057–65.
Conley A, Hinshelwood M. Mammalian aromatases. Reproduction. 2001;121:685–95.
Mehta A, Ravinder, Onteru SK, Singh D. HDAC inhibitor prevents LPS mediated inhibition of CYP19A1 expression and 17beta-estradiol production in granulosa cells. Mol Cell Endocrinol. 2015;414:73–81.
Li Q, Du X, Pan Z, Zhang L, Li Q. The transcription factor SMAD4 and miR-10b contribute to E2 release and cell apoptosis in ovarian granulosa cells by targeting CYP19A1. Mol Cell Endocrinol. 2018;476:84–95.
He W, Tao W, Zhang F, Jie Q, He Y, Zhu W, et al. Lobetyolin induces apoptosis of colon cancer cells by inhibiting glutamine metabolism. J Cell Mol Med. 2020;24:3359–69.
Tilly JL, Tilly KI, Kenton ML, Johnson AL. Expression of members of the bcl-2 gene family in the immature rat ovary: equine chorionic gonadotropin-mediated inhibition of granulosa cell apoptosis is associated with decreased bax and constitutive bcl-2 and bcl-xlong messenger ribonucleic acid levels. Endocrinology. 1995;136:232–41.
Ke F, Bouillet P, Kaufmann T, Strasser A, Kerr J, Voss AK. Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. 2013;4:e650.
Huang B, Qian C, Ding C, Meng Q, Zou Q, Li H. Fetal liver mesenchymal stem cells restore ovarian function in premature ovarian insufficiency by targeting MT1. Stem Cell Res Ther. 2019;10:362.
Lin P, Rui R. Effects of follicular size and FSH on granulosa cell apoptosis and atresia in porcine antral follicles. Mol Reprod Dev. 2010;77:670–8.
Barboni B, Turriani M, Galeati G, Spinaci M, Bacci ML, Forni M, et al. Vascular endothelial growth factor production in growing pig antral follicles. Biol Reprod. 2000;63:858–64.
Manabe N, Goto Y, Matsuda-Minehata F, Inoue N, Maeda A, Sakamaki K, et al. Regulation mechanism of selective atresia in porcine follicles: regulation of granulosa cell apoptosis during atresia. J Reprod Dev. 2004;50:493–514.
Matsuda-Minehata F, Inoue N, Goto Y, Manabe N. The regulation of ovarian granulosa cell death by pro- and anti-apoptotic molecules. J Reprod Dev. 2006;52:695–705.
Zhang LP, Song Y, Ling ZH, Li YY, Ren XY, Yang J, et al. R-spondin 2-LGR4 system regulates growth, migration and invasion, epithelial-mesenchymal transition and stem-like properties of tongue squamous cell carcinoma via Wnt/beta-catenin signaling. Ebiomedicine. 2019;44:275–88.
Du X, Li Q, Yang L, Liu L, Cao Q, Li Q. SMAD4 activates Wnt signaling pathway to inhibit granulosa cell apoptosis. Cell Death Dis. 2020;11:373.
Funding
This work was supported by China Agriculture Research System of MOF and MARA, the National Natural Science Foundation of China (31902131 and 32072694), the National Natural Science Foundation of Guangdong Province (2019A1515010676 and 2021A1515012396), the Science and Technology Project of Guangzhou (202002030071), and China Postdoctoral Science Foundation (2020M672556).
Author information
Authors and Affiliations
Contributions
X.Z., Y.H., Z.Z., H.Z., J.L. and X.Y. designed experiments. X.Z. and Y.H. performed in vivo experiments, and X.Z., Y.H., N.L. and G.B. performed in vitro experiments. X.Z. evaluated results and wrote the manuscript with revising by Y.H., X.P. and X.Y.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All experiments in the present study were performed in accordance with the guidelines of the Animal Care and Use Committee of South China Agricultural University Guangzhou, China (approval number: SYXK 2019-0136).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by B. Zhivotovsky.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, X., He, Y., Li, N. et al. DNA methylation mediated RSPO2 to promote follicular development in mammals. Cell Death Dis 12, 653 (2021). https://doi.org/10.1038/s41419-021-03941-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03941-z
