
Abstract
Background
Mutations of the PIK3CA/AKT/mTOR axis are common events in metastatic breast cancers (MBCs). This study was designed to evaluate the extent to which genetic alterations of the PIK3CA/AKT/mTOR can predict protein activation of this signalling axis in MBCs.
Methods
Molecular profiles were generated by CLIA-certified laboratories from a real-world evidence cohort of 171 MBC patients. Genetic alterations of the PIK3CA pathway were measured using next-generation sequencing. Activation levels of AKT and downstream signalling molecules were quantified using two orthogonal proteomic methods. Protein activity was correlated with underlying genomic profiles and response to CDK4/6 inhibition in combination with endocrine treatment (ET).
Results
Oncogenic alterations of the PIK3CA/AKT/PTEN pathway were identified in 49.7% of cases. Genomic profiles emerged as poor predictors of protein activity (AUC:0.69), and AKT phosphorylation levels mimicked those of mutant lesions in 76.9% of wild-type tumours. High phosphorylation levels of the PI3K/AKT/mTOR downstream target p70S6 Kinase (T389) were associated with shorter PFS in patients treated with CDK4/6 inhibitors in combination with ET (HR:4.18 95%CI:1.19–14.63); this association was not seen when patients were classified by mutational status.
Conclusions
Phosphoprotein-based measurements of drug targets and downstream substrates should be captured along with genomic information to identify MBCs driven by the PI3K/AKT/mTOR signalling.
Similar content being viewed by others


Introduction
Breast cancer remains the second-leading cause of cancer-related death for women in the United States and mortality is almost exclusively associated with the development of metastatic disease. Approximately 70% of advanced breast cancers are characterised by the expression of the oestrogen and/or progesterone hormone receptors (HR) and lack of overexpression of the human epidermal growth factor receptor 2 (HER2) [1]. The introduction of endocrine therapy (ET) in combination with a cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor has significantly impacted survival for breast cancer patients [2,3,4]. This therapeutic regimen has become the standard of care first-line treatment for newly diagnosed ER+/HER2- metastatic breast cancers (MBC). However, while this treatment has significantly improved progression-free and overall survival in MBC, most patients develop resistance.
Aberrant activation of the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR signalling pathway has been identified as a key mechanism of resistance to treatment in cancer, including ET with or without a CDK4/6 inhibitor in MBC patients [5,6,7,8,9]. Activating mutations of PIK3CA have been detected in ~40% of HR+/HER2- breast cancers, and more than 50% of breast cancer patients harbour oncogenic alterations of at least one member of the PIK3CA pathway [10, 11]. As a result, selective inhibitors aiming at modulating the activity of signalling proteins belonging to the PI3K/AKT/mTOR axis have received regulatory approval as treatment options in the MBC setting.
The mTOR complex-1 inhibitor everolimus, a targeted agent that specifically interferes with the activation of a critical downstream node of the PI3K/AKT signalling axis, was approved in 2012 in combination with exemestane for its superior efficacy in MBC patients with progressive disease compared to ET alone regardless of patients’ PIK3CA mutations status [12,13,14]. The alpha-selective PI3K inhibitor alpelisib was also recently approved as a therapeutic option in combination with fulvestrant in MBC patients whose tumours harbour oncogenic alterations of the PIK3CA gene, including patients that have progressed after receiving ET in combination with a CDK4/6 inhibitor [15, 16]. However, preclinical work by Palafox et al. has suggested that PI3K inhibition in tumours with acquired resistance to ribociclib can be independent of the PIK3CA mutation status of the tumour [5].
Based on the outcome of the randomised phase 3 CAPItello-291 trial, the Food and Drug Administration has recently approved capivasertib in combination with fulvestrant for MBC patients who develop disease progression to CDK4/6 inhibition in combination with ET. The approval is restricted to patients whose tumours harbour oncogenic alterations of the PIK3CA, AKT, or PTEN gene. However, an exploratory analysis of the trial suggests that capivasertib may also be effective in PIK3CA/AKT/PTEN non-altered tumours [17]. Similarly, a secondary biomarker analysis of the FAIRLANE trial assessing the efficacy of ipatasertib in combination with paclitaxel in the neoadjuvant setting in triple-negative breast cancers (TNBC) has suggested that phosphorylation levels of AKT were associated with clinical benefit even in the absence of genetic alterations of PIK3CA, AKT, or PTEN [18]. Likewise, several studies assessing the role of the AKT/mTOR axis in promoting resistance to ET in combination with a CDK4/6 inhibitor have suggested an essential role for genomic-independent activation of this signalling pathway in response to treatment [5, 6, 19]. Thus, a pressing question remains unanswered in the clinic: should agents targeting signalling molecules of the AKT/mTOR axis, like alpelisib and capivasertib, be solely administered to patients whose tumours harbour genomic alterations of genes encoding for members of this pathway? To answer this question, we have conducted an observational retrospective study using a real-world evidence (RWE) cohort of MBC patients and have correlated activation levels of signalling molecules of the AKT/mTOR axis with underlying genomic alterations of members of the PIK3CA pathway using a proteogenomic approach. To increase the translational potential of our work, genomic and phospho-proteomic profiles were generated by commercial and accredited laboratories.
Methods
Patients’ enrolment and samples’ collection
A retrospective cohort of 176 MBC patients was analysed from Perthera’s RWE database, a platform where patients across the United States are registered on an IRB-approved observational protocol (WCG IRB Protocol ID: PCT-01-012) and encouraged to undergo multi-omic profiling including a comprehensive next-generation sequencing (NGS) testing panel using a tumour sample at a Clinical Laboratory Improvement Amendments certified (CLIA)/College of American Pathologists (CAP)-accredited commercial laboratory. Patients entered the study through physician referrals directly to Perthera (McLean, VA) or via the Side-Out Foundation MCB programme, a patient-centred initiative sponsored by the Side-Out Foundation and contracted to Perthera. Clinical features and treatment outcomes were manually curated from physician’s notes, pathology and radiology reports and other medical records obtained via periodic records requests to patients’ treating institutions [20,21,22]. HR and HER2 status were assessed as a part of the routine care by the treating physicians and collected along with other clinical-pathological information. Patients entered the study voluntarily and each participant provided informed consent. The study was conducted following the Declaration of Helsinki guidelines. Patients with a confirmed diagnosis of MBC aged 18 years or older were eligible to enter the study.
Molecular testing was performed on Formalin-Fixed Paraffin-Embedded biospecimens collected at time of study enrolment or on archived tissue samples from surgical resections, core needle biopsies and fine needle aspirates. Generally, biopsies acquired within a year of testing were utilised for molecular profiling. Tissue sections were prepared, randomised and sent to CAP/CLIA-accredited laboratories for genomic and proteomic testing.
Next-generation sequencing
Commercial laboratories performed genomic testing for the presence and significance of pathogenic variants. The majority of NGS results were reported by Foundation Medicine (n = 78). Other comprehensive NGS panels with at least 150 genes were also generated by Caris Life Science (n = 24), Personal Genome Diagnostics (PGDx) (n = 7), Tempus (n = 11), other platforms (n = 12) and unknown (n = 31) (Fig. S1 for more information on overlapping panels). NGS-based results were harmonised into a structured format and reviewed within Perthera’s Virtual Molecular Tumour Board alongside patients’ medical and treatment histories [20].
Mutation frequency analyses were performed at the individual gene- and PI3K/AKT/mTOR pathway level. Genes included in the pathway-level analysis were identified using the Pathcards database [23]. To assess whether mutation rates in the Side-Out cohort matched those of previously analysed datasets, our findings were compared against two publicly available MBC cohorts retrieved from the Genomics Evidence Neoplasia Information Exchange (GENIE) [24,25,26,27,28]. The Inserm cohort included whole exome sequencing data collected from 216 MBC patients [27]. The Memorial Sloan Kettering Cancer Center (MSK) cohort included targeted exome sequencing data collected from 1116 individual HR+/HER2- MBC patients; data were consolidated as a single entry for patients with multiple biopsies (n = 195) [28]. Pathogenic alterations in the GENIE cohorts were determined using the Ensembl calculated variant consequences guide [29].
Immunohistochemistry
Quantification of phosphorylated AKT (pAKT) was performed on 32 FPPE samples using immunohistochemistry (IHC). Levels of pAKT were measured by Neogenomics using the Leica antibody clone LP18. Samples were scored on a dichotomous scale (high/low) and samples with staining intensities of 1+ in at least 50% of tumour cells were classified as pAKT high/positive.
Reverse phase protein microarray
Activation levels of six signalling molecules belonging to the AKT/mTOR signalling pathway were quantified in 69 patients using the Reverse Phase Protein Microarray (RPPA) assay performed by the CLIA-certified commercial laboratory Theralink Technologies. Pure tumour epithelia were isolated from the surrounding cells using laser capture microdissection, and arrays were constructed and processed as previously described [30, 31]. Primary antibodies were used to recognise the following phosphoproteins: AKT (S473) and (T308), mTOR (S2448), 4EBP1 (S65), p70S6 Kinase (T389) and S6 Ribosomal Protein (S6RP) (S235/236) (Cell Signalling, CST4060, CST4056, CST5536, CST13443, CST9234 and CST4856). Samples were analysed as previously described [32]. Normalised intensity values of each sample were fit to an analyte-specific reference standard curve. The resulting values were compared to a breast cancer reference population to determine each patient’s sample percentile score.
Breast cancer cell line subculture and treatment
Two commercially available HR+/HER2- breast cell lines, namely T47D and MCF-7, were obtained from the American Type Culture Collection (ATCC) and cultured in medium (RPMI-1640 and Eagle’s MEM, respectively) supplemented with 10% foetal bovine serum (ATCC) and human recombinant insulin in zinc solution (Thermo Fisher Scientific), per manufacturer’s instructions. Abemaciclib-resistant T47D models were generated by exposing cells to increasing amounts of the inhibitor. The IC50 concentration of abemaciclib (Selleckchem) (0.2 μM; data not shown) was used as a starting point and the amount of drug was doubled every 2 weeks to a final concentration of 2 μM, as previously described [33,34,35]. Cells were plated in a 96-well plate (Corning) and treated in technical replicates (n = 4) when they reached 80% confluency with serial dilutions of the pan-AKT inhibitor capivasertib (range 0.01–10 μM) and the PI3K inhibitor buparlisib (range 0.007–1 μM) (Selleckchem). Drug testing was performed with cells at passages 7 or 8. Cell viability was assessed 72 h after treatment using the commercially available CellTiter-Glo assay (Promega) [36]. IC50 values were calculated using a four-parameter curve fit method using GraphPad v9.5.1. For each data point, DMSO-normalised cell viability data were displayed along with standard errors of the mean.
Statistical analysis
Python’s Pandas and R v4.1.3 packages were used to perform data parsing and analyses of clinical, pathological and molecular data. Packed bubble graphs generated in Tableau v2.1 were used to display gene frequencies. The chi-square test of independence and Fisher’s exact tests were performed in the native stats R package v4.1.3 to understand relationships between categorical variables. The most appropriate test was selected based on the number of counts in each comparison group. Frequencies were displayed using mosaic plots generated with ggplot, ggmosaic, tidyverse, ggpubr and devEMF packages in R. Matrices of genomic alterations were generated in Python and data were visualised in Matlab vR2023b. Unsupervised hierarchical clustering of the RPPA continuous data was performed in Jmp Pro v17.1.0 using Ward’s method. Receiver operating characteristic (ROC) curves and area under the curve (AUC) were calculated using SPSS v28. Non-parametric Mann-Whitney and Kruskal-Wallis tests were performed in Prism v10.0.0, and continuous data were displayed as violin plots. Python Packages like Pandas, Seaborn, Matplotlib were used for data visualisation. Survival analysis and Kaplan-Meier curves were generated in R using the survival v3.5.5 and survminer v0.4.9 packages. The alpha level of significance for all comparisons was set at 0.05.
Results
Cohort description and patients’ clinical-pathological characteristics
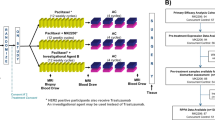
Between 2019 and 2023, 176 patients were enroled in the Side-Out initiative powered by Perthera. Patients were referred by NCI-designated cancer centres in academic institutions (NCI-CCC) (n = 65) as well as community hospitals and large non-NCI-CCC academic centres (n = 111). A CONSORT diagram describing the study population is provided in Fig. S2. Genomic profiles were generated for 171 patients enroled in the study, and phospho-proteomic data were collected by two independent laboratories, Neogenomics and Theralink Technologies, for 32 and 69 patients, respectively (Fig. 1a). An overview of the overall study design and approach can be found in Fig. 1b.

Summary of the molecular information collected from each of the 176 patients enroled in the study (a). Overall study design along with the molecular information available for each cohort and data processing pipeline (b). Packed bubble graph depicting the most frequent genomic alterations identified across the 176 patients in the Side-Out cohort (c); the dimension of each circle is proportional to the number of occurrences. Red circles indicate genes detected in at least 10% of the study population. Matrix illustrating frequency and co-occurrences of the ten most frequently mutated or amplified genes in the Side-Out cohort (d and e, respectively).
The median age of participants at diagnosis was 59 (range 28–85), and the cohort included 173 females and three males (Table 1). Race and ethnicity information were available for 119 patients, of which 96 were White (80.7%), 12 were Black (10.1%), seven were Asians (5.9%) and four were Hispanic (3.4%) (Table 1).
Of the 176 participants, 110 (62.5%) were affected by HR+/HER2- tumours, 48 (27.3%) by TNBCs, 13 (7.4%) by HER2+ tumours, including nine HR+/HER2+ (5.1%) and four by HER2-enriched (HR-/HER2+) (2.3%) lesions (Table 1).
Of the 176 patients, 65.3% (n = 115) had stage 4 metastatic disease at the time of diagnosis. The liver (n = 27, 25.0%) was the most frequent site of metastasis, and 23 patients (21.3%) had secondary lesions at more than one site (Table 1). The number of treatments before study participation ranges from one to six (Table 1).
The PIK3CA pathway mutational landscape in a real-world evidence cohort of metastatic breast cancer patients mimics those of well-characterised cohorts
We first assessed the frequencies of the most common genomic alterations in the Side-Out cohort of MBC patients (Fig. 1c; Fig. S3; Fig. S4; Table S1). For nine of 171 patients (5.3%) with genomic data, none of the genes measured by NGS harboured oncogenic alterations (Fig. 1d). As expected, TP53 (n = 75, 43.9%) and PIK3CA (n = 58, 33.9%) mutations were the most frequently identified pathogenic variants (Fig. 1d), followed by amplifications of FGF3 (n = 24, 14.0%), FGF4 (n = 23, 13.5%), CCND1 (n = 22, 12.9%), MYC (n = 21, 12.3%) and mutations of ESR1 (n = 21, 12.3%) and GATA3 (n = 19, 11.1%) (Fig. 1d, e). The complete genomic landscape of the entire cohort can be found in Fig. S3 and S4.
PIK3CA alterations were found in 43.5% of HR+/HER2- tumours (n = 47), 50% of HER2-enriched lesions (n = 2), 33.3% of HR+/HER2+ (n = 3) and 11.1% of TNBCs (n = 5) (Fig. 2a). The three most frequent PIK3CA gain-of-function pathogenic variants were the H1047R (n = 26, 15.2%) mutation of the kinase domain, and the E545K (n = 15, 8.8%) and E542K (n = 6, 3.5%) mutations of the helical domain (Fig. 2a). Within the same sample, multiple pathogenic alterations of the PIK3CA gene were found in one TNBC (2%) and 10 HR+/HER2- tumours (9%) (Fig. 2a). PIK3CA amplifications were found in ~2% (n = 3) of patients including two patients with HR+/HER2- MBCs and one patient with a TNBC tumour. PIK3CA amplifications were exclusively found in tumours that harboured PIK3CA gain-of-function mutations affecting the H1047R and E542K residues. Genetic alterations of the PIK3CA gene co-occurred with pathogenic alterations of ESR1 and TP53 in 7% (n = 12) and 10.5% (n = 18) of patients, respectively (Fig. 1d).

Matrix illustrating frequencies of mutations and amplifications across ten members of the PIK3CA pathway in the Side-Out cohort (a); tumours were subdivided based on the molecular subtypes. HR+/HER2- tumours are shown in the top panel (a). Cumulative number of cases with genetic alterations of members of the PIK3CA pathway by tumour subtype in the Side-Out cohort (b) and the Inserm cohort retrieved from the GENIE database (c). Matrix illustrating frequencies of mutations and amplifications across ten members of the PIK3CA pathway in the Inserm cohort (d); tumours were subdivided based on the molecular subtypes; HR+/HER2- tumours are shown in the top panel.
Given the primary role of the PI3K/AKT signalling pathway in breast cancer onset and progression, especially in HR+/HER2- tumours, we next extended our analysis to other genes encoding for members of this signalling axis. Of the 92 genes listed in the Pathcards database as members of the PIK3CA/AKT ‘SuperPath’ [23], oncogenic alternations of 12 genes, namely PIK3CA, AKT 1, 2 and 3, PTEN, MDM2, PIK3R1, RICTOR, RPTOR, FOXO1, FOXO3 and SGK1, were identified in our cohort of patients (Fig. 2a, b).
Oncogenic alterations of genes belonging to the PIK3CA/AKT ‘SuperPath’ were identified in 85 (49.7%) of the 171 patients analysed (Fig. 2a). Sixteen of the 171 patients (9.4%) had aberrations of more than one gene belonging to the PIK3CA pathway (Fig. 2a). Oncogenic alterations of AKT1, 2 and 3 were found in 16 patients (9.4%), including nine AKT1 E17K point mutations, five AKT3 and two AKT2 amplifications (Figs. 1d, 2a). Of interest, PIK3CA oncogenic alterations were found in five of the seven patients with amplified AKT2 or AKT3. Loss-of-function alterations of the tumour suppressor PTEN were found in 15 patients (8.8%) and co-occurred with PIK3CA mutations in five patients. Additional alterations of members of the PIK3CA/AKT pathway included mutations of MDM2 in five patients (2.9%) and of PIK3R1 and RICTOR in two (1.2%) patients. Oncogenic mutations of FOXO1, FOXO3, RPTOR and SGK1 were found in one patient each across the entire cohort (0.6%, respectively) (Fig. 2a).
We next compared mutation rates of members of the PIK3CA pathway and their distribution in the Side-Out cohort against two publicly available MBC datasets retrieved from the GENIE database, the Inserm and the MSK cohort (Fig. 1b). While the MSK cohort only included HR+/HER2- tumours (n = 1116), biospecimens in the Inserm cohort (n = 216) contained all major breast cancer subtypes including 143 (66.2%) HR+/HER2-, 51 (23.6%) TNBC and 14 (6.5%) HER2-enriched MBCs. The molecular subtype of the remaining eight tumours was unknown.
PIK3CA pathogenic alterations were found in 31.5% (n = 68) of patients in the Inserm cohort and 40.1% (n = 448) in the MSK cohort (p = 0.03) (Fig. 2c, d, Fig. S5, Table 2). When the analysis was restricted to HR+/HER2- tumours, no differences were detected across the three cohorts (p > 0.05; Table 2).
In the Inserm cohort, PIK3CA oncogenic alterations were detected in 55 (38.5%) HR+/HER2- tumours, 6 (11.8%) TNBC, 3 (21.4%) HER2-enriched lesions, four tumours with unknown subtype (Fig. 2c, d). Amplification of the PIK3CA gene was observed in 5 HR+/HER2- tumours (3.5%) and one TNBC patient (2%). When the analysis was extended to the other members of the PIK3CA/AKT ‘SuperPath,’ members of the PIK3CA pathway were found to be altered in 127 patients (58.8%) in the Inserm cohort. Oncogenic alterations of more than one gene were found in 45 biospecimens (Fig. 2d). AKT alterations were detected in 34 (15.7%) patients. PTEN mutations were present in 14 (6.5%) cases (Fig. 2d). Mutations of PIK3R1 were found in 16 (7.4%) individuals and of RICTOR and RPTOR in 13 (6%) and 12 (5.6%) patients, respectively (Fig. 2d). Pathogenic alterations of the transcription factors FOXO1 and FOXO3 were found in four (1.9%) and eight (3.7%) patients, respectively.
In the MSK cohort, oncogenic aberrations of the PIK3CA pathway were detected in 663 patients (59.4%), of which 148 had alterations of multiple genes. AKT was altered in 131 (11.7%) patients and PTEN in 95 (8.5%) patients (Fig. S3). Mutations of PIK3R1 were found in 26 (2.3%) patients and of RICTOR (2.2%) and RPTOR (2.2%) in 25 patients, respectively. Frequencies of genomic alterations of the main members of the PIK3CA pathway were at large similar among the three cohorts, with the exception of a few genes that were relatively infrequent across study sets and of PIK3CA when the analysis was not restricted to the HR+/HER2- tumours, which is the only subtype represented in the MSK cohort (Table 2).
AKT functional activation in metastatic breast cancers cannot be solely predicted by the underlying genomic profile
Next, we assessed whether the underlying genomic profile could predict the activation of critical nodes on the PI3K/AKT/mTOR signalling axis. We used two orthogonal proteomic methods to determine the association between genomic alterations and activation of the PIK3CA/AKT/mTOR pathway with standard IHC and RPPA.
We first assessed whether AKT (pAKT) activation was more frequently detected in tumours harbouring genetic alterations of the PIK3CA/AKT/mTOR axis compared to wild-type lesions using the IHC data. Of the 32 samples analysed, 25 had high activation of pAKT. The proportion of patients with high pAKT activity was similar in tumours harbouring pathogenic alterations of any member of the PIK3CA pathway compared to wild-type tumours (79% versus 77%, respectively; p > 0.05) (Fig. 3a). Approximately three quarters (76.9%) of PIK3CA wild-type tumours had pAKT levels that mimicked those of mutant lesions suggesting that AKT activity in MBCs is driven by genomic-dependent and independent events (Fig. 3b). Similar associations were also found when the analysis was restricted to the aberration of PIK3CA, AKT and PTEN, individually (Fig. 3a).

Mosaic plots showing correlations between mutational status and phosphorylated levels of AKT (pAKT) measured by IHC and classified on a dichotomous scale (high versus low) (a). The proportion of patients with high AKT activity was first compared between wild-type and PIK3CA, AKT and PTEN mutant/amplified tumours. The analysis was then extended to compare AKT activity in wild-type tumours and lesions with any genetic alterations of the PIK3CA pathway (PIK3CA pathway alterations). Tile plots summarising frequencies of genetic alterations of the PIK3CA pathway along with phosphorylated AKT levels measured by IHC (b). Unsupervised hierarchical clustering Ward’s method assessing activation of six members of the PI3K/AKT/mTOR signalling axis in wild-type tumours and lesions harbouring oncogenic alterations of the PIK3CA pathway (c). Functional protein activation was measured on a continuous scale using RPPA percentile scores. Tumours’ molecular subtypes are listed and colour-coded on the x-axis. On the y-axis, samples with underlying genomic alterations are shown in black. RPPA continuous data are shown on a blue (low activation) to red (high activation) scale. Violin plots comparing activation of the signalling molecules in samples harbouring alterations of any gene of the PIK3CA axis and wild-type tumours (d); sample median is shown for each plot and asterisks denote comparisons that were statistically different (p < 0.01). The receiver operating characteristic (ROC) curve shows the performance of mutations of the PIK3CA pathway as potential classifiers for predicting AKT phosphorylation levels in MBCs along with the corresponding area under the curve (AUC) (e). Violin plots comparing activation levels of signalling molecules across the three most frequently detected PIK3CA oncogenic mutations (f).
We next compared activation levels of six proteins involved in the PI3K/AKT/mTOR signalling axis measured by RPPA on a continuous scale; RPPA values were calculated as percentile scores of the reference population [37]. We first used unsupervised hierarchical clustering to examine how key proteins belonging to the PI3K/AKT/mTOR signalling axis were distributed in 64 samples for which RPPA and NGS data were available (Fig. 3c). At large, subtypes were not associated with AKT/mTOR activation (Fig. 3c, Fig. S6). Samples harbouring mutations of genes encoding for proteins belonging to the PI3K/AKT/mTOR axis were spread across three clusters and had heterogeneous activation of the signalling molecules. Cluster 1 was characterised by increased activation of mTOR and its downstream signalling molecules. Of the 29 patients in this cluster, 15 had genomic alterations of target genes. Cluster 2 was characterised by higher activation levels of AKT and its downstream targets and contained ten samples, including three of the eight tumours harbouring PTEN loss of function mutations. Samples included in Cluster 3 had an overall low activation of the different signalling molecules regardless of the mutational status.
When tumours with any mutation of the PIK3CA pathway were compared to wild-type lesions, activation of AKT (T308) and (S473) was significantly higher in the mutant population (p < 0.01). However, a significant degree of overlap in the samples’ distribution was detected between the two groups (Fig. 3d). ROC analysis assessing the ability of the mutational status to predict AKT activation suggested that pathogenic alterations of the PIK3CA pathway are poor predictors of protein activity (AUC: 0.69; Fig. 3e). When the analysis was restricted to PIK3CA, AKT and PTEN individually, activation of the signalling molecules was not different between wild-type and mutant tumours (p > 0.05; data not shown). Likewise, when a pathway score was created using the RPPA data as previously described [18], activation of the signalling molecules by RPPA did not differ between the mutant and wild-type populations (data not shown). Similar trends were also observed when the analysis was restricted to the HR+/HER2- population (Fig. 4a, b). Lastly, activation of signalling molecules did not differ between tumours harbouring different PIK3CA pathogenic mutations (Fig. 3f) nor in tumours harbouring multiple pathogenic alterations of genes belonging to the PIK3CA pathway (Fig. S7).

Violin plots comparing activation levels, measured as RPPA percentile scores, of signalling molecules in HR+/HER2- MBCs harbouring alterations of any gene of the PIK3CA axis (a); sample median is shown for each plot, and asterisks denote comparisons that were statistically different (p < 0.02). The receiver operating characteristic (ROC) curve shows the performance of mutations of the PIK3CA pathway as potential classifiers for predicting AKT phosphorylation levels in HR+/HER2- MBCs along with the corresponding area under the curve (AUC) (b). Heat map capturing activation levels of AKT (S473) and (T308) across 29 cell lines publicly available in the DepMap database. HR+/Her2- cells are shown in blue (c). Heat map showing PIK3CA dependency in 38 cell lines that underwent CRISPR and Ribonucleic acid interference (RNAi) screening (d). Unsupervised hierarchical clustering using Ward’s method assessing activation of the PI3K/AKT/mTOR signalling axis in the MCF-7, T47D and T47D abemaciclib-resistant cells; functional protein activation was measured on a continuous scale using RPPA values (e). Cell viability line plot of MCF-7 and T47D cells treated with capivasertib (range from 0.01–10 μM) along with box plot depicting the RPPA continuous values of phospho-AKT (T308) and phospho-4EBP1 (S65) and (T70) (p < 0.01). Median, highest and lowest values of experimental replicates are shown; asterisks denote comparisons that are statistically significant (f). Cell viability line plot of T47D parental and abemaciclib-resistant cells treated with buparlisib (range from 0.007 to 1 μM) along with box plot depicting the RPPA continuous values of phospho-AKT (T308) and phospho-p70S6 Kinase (T389) (p < 0.01 and 0.05, respectively). Medians, highest and lowest values of experimental replicates are shown; the asterisks denote comparisons that are statistically significant (g). Kaplan-Meier plot along with hazard ratio for progression-free survival in days and 95% confidence interval for patients with genomic alterations of any members of the PIK3CA pathway (HR: 0.58; CI: 0.14–2.36), PIK3CA (HR:0.67; CI 0.19–2.38), and phospho-p70 S6 Kinase (T389) activity (HR: 4.18; CI: 1.19–14.63) (h). P70S6 kinase activity levels were classified on a binary scale (high/low) based on the population median of the continuous RPPA data. Diagram showing a workflow for integrating multi-omic-based profiling for allocating patients to targeted treatments against members of the PI3K/AKT/mTOR pathway (i).
We next assessed whether PIK3CA mutations predicted AKT activation in 29 breast cancer cell lines retreated from the publicly available DepMap portal. At large, activation of AKT was not higher in cell lines harbouring a PIK3CA oncogenic mutation (Fig. 4c). We next looked at the significance of PIK3CA in relation to the lethality of its deletion or knockdown on a target across all 38 cell lines included in the dataset. Negative dependence scores suggest cell lines are heavily dependent on that gene. In line with our protein data, only two cell lines of the PIK3CA mutant cell lines, MDAMB361 and EFM19, showed dependency from the PI3K (Fig. 4d). To confirm the role of signalling molecules in predicting response to agents targeting the PI3K/AKT/mTOR pathway, we next compared the sensitivity to capivasertib in two HR+/HER2- breast cancer cell lines, namely T47D and MCF-7, harbouring hotspot mutations of PIK3CA (H1047R and E545K, respectively) [38] with diverse levels of activation of the PI3K/AKT/mTOR axis (Fig. 4e). Compared to the MCF-7, T47D cells had lower IC50 values (2.1 vs. 7.5 μM) and higher levels of phosphorylated AKT (T308) and 4EBP1 (S65) and (T70) (Fig. 4f). Given the increased levels of AKT activity in the T47D cell lines compared to the MCF-7 cells, we next assessed responses to the PI3K inhibitor buparlisib in parental and isogenic abemaciclib-resistant cells. As expected, resistant cells were more sensitive to buparlisib and increased sensitivity was associated with higher levels of phosphorylation of AKT (T308) as well as increased activation of the downstream substrate p70S6 Kinase (Fig. 4g).
Taken together, these data suggest that inferring activation of signalling molecules merely by the underlying genomic alterations of the PIK3CA pathway underestimates the activation frequencies of the PI3K/AKT/mTOR axis in MBCs. As AKT and PI3K inhibitors are currently being used in the clinical setting, capturing activity levels of signalling molecules belonging to this axis may help identify MBCs driven by the PI3K/AKT/mTOR signalling pathway and thus may benefit from these treatments.
Functional activation of AKT downstream substrates is associated with response to first-line treatment in HR+/HER2- metastatic breast cancers
As inhibition of AKT and PI3K activity has recently been shown to delay resistance to ET in combination with a CDK4/6 inhibitor in MBCs in preclinical studies [5, 19], we next assessed whether baseline activation levels of members of the PI3K/AKT/mTOR pathway are associated with response to first-line treatment with a CDK4/6 inhibitor in the Side-Out cohort. Of the 69 patients for which RPPA data were collected, 20 were treated with a CDK4/6 inhibitor in combination with ET in first-line. NGS data were available for 18 of the 20 patients included in this sub-analysis (Table S3). As expected, the mutation status of the PIK3CA pathway or PIK3CA was not associated with progression-free survival (PFS) (p > 0.05) (Fig. 4h). However, patients with p70S6 Kinase (T389) levels above the whole population median had shorter PFS compared to those with low p70S6 Kinase (T389) activity (HR: 4.18, 95% CI:1.19–14.63, p = 0.02; Fig. 4h).
Taken together, our data suggest that genomic alterations of the PIK3CA pathway are insufficient to predict protein activation in clinical samples and patients’ response to standard of care.
Discussion
Using a RWE cohort of patients, for which broad molecular profiles were collected in commercial and certified laboratories [20,21,22], we have conducted a comprehensive analysis to assess the effects of genetic alterations of the PIK3CA pathway on PI3K/AKT/mTOR activity in MBCs. Our data suggest that the Side-Out cohort analysed as part of this study has PIK3CA genomic pathway profiles similar to those of previously analysed cohorts enroled in elective centres [10, 11, 24,25,26, 39]. As expected, PIK3CA mutations were amongst the most frequently detected pathogenic variants with 33.9% of all tumours and 43.5% of HR+/HER2- MBCs harbouring oncogenic alterations of this key signalling hub [39]. Hotspot mutations on exon 9 (E545K and E542K) and 20 (H1047R) were found in 81% of the mutant lesions, with the H1047R mutation of the kinase domain being the most frequently detected alteration across the 58 mutant specimens (44.8%) [40]. Because most of the genetic alterations detected in our cohort of patients are known to translate into PI3K gain-of-function activity, we next used our sample set to understand the effects these genetic alterations exert on the PI3K/AKT/mTOR signalling axis and the extent to which they can be used to predict pathway activation.
Whether the activation status of AKT was determined using IHC or RPPA-based measurements, genetic alterations of PIK3CA and other members of this pathway emerged as poor predictors of protein activity across MBCs. Similarly, a recent analysis assessing AKT activation and its association with PIK3CA status in MBCs conducted by Alves et al. has shown that phosphorylation levels of AKT measured by IHC were independent of PIK3CA mutational status as high and low AKT levels were found in both mutant and wild-type tumours [19]. Likewise, a secondary biomarker analysis of the FAIRLANE trial, where AKT activity was measured by RPPA and dichotomised based on the population median, showed that 44% of samples with low AKT activity had pathogenic alterations of PIK3CA/AKT1/PTEN by NGS or IHC [18]. As the PI3K/AKT/mTOR signalling axis is a central hub where many signal transduction pathways are integrated [41], it is not surprising that the mutational status of the PIK3CA axis alone is often insufficient to fully capture the activation status of the PI3K/AKT/mTOR signalling axis.
Over the years, targeting malfunctioning PI3K/AKT/mTOR has been considered an attractive therapeutic target, especially in the MBC metastatic setting, as activation of this signalling axis has been linked to cancer progression and metastasization [10, 42]. Several compounds targeting PI3K have been tested in the clinic, and biomarker analyses have shown that, at large, these compounds are most effective when administered to patients affected by MBC harbouring oncogenic alterations of the PIK3CA gene [43]. André and colleagues have shown prolonged PFS and increased clinical benefit in PIK3CA-mutated MBC patients treated with alpelisib in combination with fulvestrant compared to patients treated with placebo and fulvestrant [15]. However, when restricted to the PIK3CA wild-type cohort of patients, the same proof-of-concept criteria were not met [15]. Similarly, in the BELLE-2 randomised clinical trial assessing response to buparlisib in previously treated HR+/HER2- locally advanced or metastatic breast cancer patients, longer PFS was detected specifically in patients where PIK3CA mutations were detected in circulating tumour DNA (ctDNA). These differences were lost in the ctDNA non-mutant group, suggesting that PIK3CA status may be a good predictor of response to PI3K inhibitors [44].
However, this association does not appear to be as linear when targeting PI3K downstream signalling molecules [13]. Correlative biomarker analysis of the BOLERO2 trial assessing the efficacy of everolimus in combination with exemestane in HR+/HER2- refractory advanced breast cancers showed that PFS was independent of underlying genetic alteration of PIK3CA [13, 39]. Similarly, an exploratory analysis of the CAPItello-291 trial, which led to the approval of capivasertib in combination with fulvestrant for MBC patients whose tumours harbour oncogenic alterations of the PIK3CA, AKT, or PTEN gene, has suggested that capivasertib retains activity beyond AKT-pathway altered tumours [17]. This observation aligns with extensive preclinical work demonstrating that response to capivasertib in breast cancer models is highly dependent upon levels of AKT activity [45, 46] and that phosphorylation of AKT cannot be fully predicted by genomic alterations of its regulators [6, 10, 18].
Aberrant activation of this signalling axis is also a well-known resistance mechanism to various treatments, including ET and CDK4/6 inhibitors [6, 42]. Several studies have shown that inhibition of members of the PI3K/AKT/mTOR can delay the acquisition of resistance to CDK4/6 inhibitors [5, 19]. We have shown that increased activation of the PI3K/AKT/mTOR signalling axis in samples collected from MBC patients that develop disease progression when treated with a CDK4/6 inhibitor in combination with ET extends beyond cancer cells into the surrounding stroma/immune compartment [5]. This suggests that genomic-independent activation of the signalling axis may affect PI3K/AKT/mTOR activity within the tumour microenvironment as a whole [5]. In the current study, we confirm that phosphorylation of signalling molecules, a biochemical event that leads to the activation of members of the PI3K/AKT/mTOR pathway, is associated with response to CDK4/6 inhibitor in combination with ET in MBC patients [9, 47]. However, this association was lost when patients were subclassified based on the mutation status of the PIK3CA gene and other members of this signalling axis.
While our study provides strong evidence of the limited extent to which genetic alterations of the PIK3CA pathway may predict PI3K/AKT/mTOR activity in MBCs, a few limitations need to be addressed. Enrolment in the Side-Out cohort may be biased toward academically inclined physicians who are more prone to utilise molecular information in their clinical practice. However, given the broad catchment area and number of enroling institutions involved in the study, we believe this cohort is a good representation of heterogeneous populations of MBC patients. In addition, given the RWE nature of the study cohort, capturing standardised and accurate information on patients’ comorbidities and medical history outside of the cancer diagnosis can be challenging. Thus, covariates not accounted for in our study may affect patients’ performance status (and consequently eligibility for some anti-cancer treatment) and outcome, regardless of the underlying biological events driving individual tumours.
Second, tissue samples and molecular profiles were collected at different time points from each patient; thus, correlating molecular data and outcomes associated with specific treatments can be challenging in this cohort. However, this is a common issue related to many biomarker studies, and our cohort is well suited for generating new hypotheses or validating independent findings.
Lastly, as tissues were profiled with different NGS platforms (Fig. S1), the frequency of some of the alterations reported may have been underestimated. However, all platforms captured the most frequently mutated genes and most amplifications. Similarly, for genes belonging to the PIK3CA pathway, only genes with low frequencies were not measured by all panels (e.g. FOXO, RICTOR, RPTOR). While these discrepancies may affect our data, the overlap between our cohort and two independent studies suggests that even with these limitations, our work provides an accurate overview of the molecular landscape of MBCs in an RWE.
Despite these limitations, this study reinforces the need for devising multi-omic-based approaches that account for functional signalling data to be incorporated into decision-making pipelines for allocating patients to precision treatments. As the landscape of FDA-approved therapeutics continues to expand for MBC patients, especially for those affected by HR+/HER2− disease, molecularly rationalised treatment selection becomes gradually more important to effectively allocate patients to treatment. Given the recent approval of agents targeting the PI3K/AKT/mTOR axis, our data provide timely and important insights on the role of multi-omic profiling for MBC patients, even if based on an interim analysis of a growing RWE cohort. With different assays available in commercial laboratories designed to capture expression and activation levels of these drug targets and their substrates, functional data should routinely be evaluated as an integral part of a tumour’s molecular landscape to guide treatment selection for MBC patients (Fig. 4i) [20, 48, 49].
Data availability
Molecular profiles for patients in the Side-Out cohort can be accessed at https://sideoutfoundation.gmu.edu/. Data from the GENIE cohort were accessed at https://www.cbioportal.org/. CRISPR- and RNAi-based PIK3CA dependency scores and RPPA-based measurements of phosphorylated AKT (S473 and T308) for 38 cell lines were obtained from Cancer Dependency Map (DepMap) Data Explorer (https://depmap.org/).
References
Johnson KS, Conant EF, Soo MS. Molecular subtypes of breast cancer: a review for breast radiologists. J Breast Imaging. 2021;3:12–24.
Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med. 2016;375:1925–36.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35.
Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379:1926–36.
Palafox M, Monserrat L, Bellet M, Villacampa G, Gonzalez-Perez A, Oliveira M, et al. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6 inhibitor resistance in ER+ breast cancer. Nat Commun. 2022;13:5258.
Abu-Khalaf MM, Alex Hodge K, Hatzis C, Baldelli E, El Gazzah E, Valdes F, et al. AKT/mTOR signaling modulates resistance to endocrine therapy and CDK4/6 inhibition in metastatic breast cancers. Npj Precis Oncol. 2023;7:1–12.
Hanker AB, Sudhan DR, Arteaga CL. Overcoming endocrine resistance in breast cancer. Cancer Cell. 2020;37:496–513.
Tokunaga E, Kataoka A, Kimura Y, Oki E, Mashino K, Nishida K, et al. The association between Akt activation and resistance to hormone therapy in metastatic breast cancer. Eur J Cancer Oxf Engl. 2006;42:629–35.
Karlsson E, Pérez-Tenorio G, Amin R, Bostner J, Skoog L, Fornander T, et al. The mTOR effectors 4EBP1 and S6K2 are frequently coexpressed, and associated with a poor prognosis and endocrine resistance in breast cancer: a retrospective study including patients from the randomised Stockholm tamoxifen trials. Breast Cancer Res. 2013;15:R96.
Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19 784 diverse solid tumors. JAMA Oncol. 2016;2:1565–73.
Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–9.
O’Shaughnessy J, Thaddeus Beck J, Royce M. Everolimus-based combination therapies for HR+, HER2- metastatic breast cancer. Cancer Treat Rev. 2016;69:204–14.
Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol J Eur Soc Med Oncol. 2014;25:2357–62.
André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer. N Engl J Med. 2019;380:1929–40.
Rugo HS, Lerebours F, Ciruelos E, Drullinsky P, Ruiz-Borrego M, Neven P, et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021;22:489–98.
Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortes J, Gomez Moreno HL, et al. Capivasertib in hormone receptor-positive advanced breast cancer. N Engl J Med. 2023;388:2058–70.
Shi Z, Wulfkuhle J, Nowicka M, Gallagher RI, Saura C, Nuciforo PG, et al. Functional mapping of AKT signaling and biomarkers of response from the FAIRLANE trial of neoadjuvant ipatasertib plus paclitaxel for triple-negative breast cancer. Clin Cancer Res. 2022;28:993–1003.
Alves CL, Ehmsen S, Terp MG, Portman N, Tuttolomondo M, Gammelgaard OL, et al. Co-targeting CDK4/6 and AKT with endocrine therapy prevents progression in CDK4/6 inhibitor and endocrine therapy-resistant breast cancer. Nat Commun. 2021;12:5112.
Pishvaian MJ, Blais EM, Bender RJ, Rao S, Boca SM, Chung V, et al. A virtual molecular tumor board to improve efficiency and scalability of delivering precision oncology to physicians and their patients. JAMIA Open. 2019;2:505–15.
Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020;21:508–18.
Pishvaian MJ, Bender RJ, Halverson D, Rahib L, Hendifar AE, Mikhail S, et al. Molecular profiling of patients with pancreatic cancer: initial results from the know your tumor initiative. Clin Cancer Res J Am Assoc Cancer Res. 2018;24:5018–27.
Belinky F, Nativ N, Stelzer G, Zimmerman S, Iny Stein T, Safran M, et al. PathCards: multi-source consolidation of human biological pathways. Database. 2015;2015:bav006.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
de Bruijn I, Kundra R, Mastrogiacomo B, Tran TN, Sikina L, Mazor T, et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR project GENIE biopharma collaborative in cBioPortal. Cancer Res. 2023;83:3861–7.
Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, et al. Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Med. 2016;13:e1002201.
Li Q, Jiang B, Guo J, Shao H, Del Priore IS, Chang Q, et al. INK4 tumor suppressor proteins mediate resistance to CDK4/6 kinase inhibitors. Cancer Discov. 2022;12:356–71.
Martin FJ, Amode MR, Aneja A, Austine-Orimoloye O, Azov AG, Barnes I, et al. Ensembl 2023. Nucleic Acids Res. 2023;51:D933–41.
Gallagher RI, Espina V. Reverse phase protein arrays: mapping the path towards personalized medicine. Mol Diagn Ther. 2014;18:619–30.
Ingham M, Lee S, Van Tine BA, Choy E, Oza J, Doshi S, et al. A single-arm phase II trial of sitravatinib in advanced well-differentiated/dedifferentiated liposarcoma. Clin Cancer Res J Am Assoc Cancer Res. 2023;29:1031–9.
Baldelli E, Hodge KA, Bellezza G, Shah NJ, Gambara G, Sidoni A, et al. PD-L1 quantification across tumor types using the reverse phase protein microarray: implications for precision medicine. J Immunother Cancer. 2021;9:e002179.
Pandey K, Katuwal NB, Park N, Hur J, Cho YB, Kim SK, et al. Combination of abemaciclib following eribulin overcomes palbociclib-resistant breast cancer by inhibiting the G2/M cell cycle phase. Cancers. 2022;14:210.
Ono M, Oba T, Shibata T, Ito KI. The mechanisms involved in the resistance of estrogen receptor-positive breast cancer cells to palbociclib are multiple and change over time. J Cancer Res Clin Oncol. 2021;147:3211–24.
Pancholi S, Ribas R, Simigdala N, Schuster E, Nikitorowicz-Buniak J, Ressa A, et al. Tumour kinome re-wiring governs resistance to palbociclib in oestrogen receptor positive breast cancers, highlighting new therapeutic modalities. Oncogene. 2020;39:4781–97.
Baldelli E, El Gazzah E, Moran JC, Hodge KA, Manojlovic Z, Bassiouni R, et al. Wild-type KRAS allele effects on druggable targets in KRAS mutant lung adenocarcinomas. Genes. 2021;12:1402.
Baldelli E, Calvert V, Hodge A, VanMeter A, Petricoin EF, Pierobon M. Reverse phase protein microarrays. Methods Mol Biol. 2017;1606:149–16.
Sondka, Dhir Z, Carvalho-Silva NB, Jupe D, Madhumita null S, McLaren K, et al. COSMIC: a curated database of somatic variants and clinical data for cancer. Nucleic Acids Res. 2024;52:D1210–7.
Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA, Pritchard KI, et al. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor–positive, human epidermal growth factor receptor 2–negative advanced breast cancer: results from BOLERO-2. J Clin Oncol. 2016;34:419–26.
Martínez-Sáez O, Chic N, Pascual T, Adamo B, Vidal M, González-Farré B, et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020;22:45.
Mahajan K, Mahajan NP. PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J Cell Physiol. 2012;227:3178–84.
Beelen K, Hoefnagel LD, Opdam M, Wesseling J, Sanders J, Vincent AD, et al. PI3K/AKT/mTOR pathway activation in primary and corresponding metastatic breast tumors after adjuvant endocrine therapy. Int J Cancer J Int Cancer. 2014;135:1257–63.
Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273–91.
Baselga J, Im SA, Iwata H, Cortés J, De Laurentiis M, Jiang Z, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:904–16.
Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11:873–87.
Gris-Oliver A, Palafox M, Monserrat L, Brasó-Maristany F, Òdena A, Sánchez-Guixé M, et al. Genetic alterations in the PI3K/AKT pathway and baseline AKT activity define AKT inhibitor sensitivity in breast cancer patient-derived xenografts. Clin Cancer Res J Am Assoc Cancer Res. 2020;26:3720–31.
Beelen K, Opdam M, Severson TM, Koornstra RH, Vincent AD, Wesseling J, et al. Phosphorylated p-70S6K predicts tamoxifen resistance in postmenopausal breast cancer patients randomized between adjuvant tamoxifen versus no systemic treatment. Breast Cancer Res. 2014;16:R6.
Pierobon M, Robert NJ, Northfelt DW, Jahanzeb M, Wong S, Hodge KA, et al. Multi-omic molecular profiling guide’s efficacious treatment selection in refractory metastatic breast cancer: a prospective phase II clinical trial. Mol Oncol. 2022;16:104–15.
Randall J, Hunt AL, Nutcharoen A, Johnston L, Chouraichi S, Wang H, et al. Quantitative proteomic analysis of HER2 protein expression in PDAC tumors. Clin Proteom. 2024;21:24.
Acknowledgements
We thank all the patients who participated in the study and their families.
Funding
This project was fully funded by the Side-Out Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualisation: EMB, QW and MP; Methodology: DP, EB, EMB, JD, EEG, CM, AG, AI, AVN and BAC; Investigation: DP, EB, EMB, JD, EEG, CM, AG, AI, AVN, BAC, EFP, QW and MP; Visualisation: DP, EB, QW and MP; Funding acquisition: RD; Supervision: QW and MP; Writing—original draft: DP, EB and MP; Writing—review & editing: DP, EMB, JD, EEG, CM, AG, AI, AVN, BAC, RD, EFP and QW.
Corresponding author
Ethics declarations
Competing interests
The authors are inventors of US Government and University-assigned patents and patent applications covering aspects of the technologies. As inventors, they are entitled to receive royalties as provided by US Law and George Mason University policy. MP and EFP receive royalties from Theralink Technologies, Inc. MP and EFP are consultants to and/or shareholders of Theralink Technologies, Inc; EFP is a shareholder and consultant of Perthera, Inc.
Ethics approval and consent to participate
The retrospective cohort of 176 metastatic breast cancer patients was analysed from Perthera’s platform, where patients are registered on an IRB-approved observational protocol (WCG IRB Protocol ID: PCT-01-012). Patients entered the study voluntarily, and each participant provided informed consent. The study was conducted following the Declaration of Helsinki guidelines.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prasad, D., Baldelli, E., Blais, E.M. et al. Functional activation of the AKT-mTOR signalling axis in a real-world metastatic breast cancer cohort. Br J Cancer (2024). https://doi.org/10.1038/s41416-024-02852-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41416-024-02852-y
