
Abstract
Vitamin D analogs have been widely utilized for the treatment of vitiligo, but the molecular mechanism underlying their pharmacological effects (especially their antioxidant properties) has not yet been investigated. We evaluated the relationship between serum vitamin D level and oxidative damage severity in vitiligo patients, and investigated the molecular mechanism of vitamin D in protecting melanocytes against oxidative stress. Serum levels of 25-hydroxyvitamin D and malondialdehyde (MDA) were first measured in patients. A variety of in vitro experiments such as intracellular reactive oxygen species (ROS), cellular viability, migration, and apoptotic assays were then performed to detect the effects of vitamin D or β-catenin silencing on H2O2-treated melanocytes. Expression of Wnt/β-catenin, Nrf2, apoptotic, and MITF pathways was finally examined using quantitative real-time PCR and western blot. In this study, we initially found that vitamin D insufficiency was closely associated with the severity of oxidative stress in vitiligo patients. Using ex vivo cell models, we further showed that vitamin D positively modulated β-catenin signaling at both translational and posttranslational levels in melanocytes under oxidative stress. Like WNT agonists, vitamin D significantly inhibited ROS accumulation and cell apoptosis in H2O2-treated melanocytes and promoted their proliferative and migratory activity, while the protective effects of vitamin D against oxidative stress were abolished by β-catenin silencing in melanocytes. Furthermore, β-catenin deficiency also blocked the activation of Nrf2 and MITF as well as the inhibition of apoptosis induced by vitamin D. Taken together, vitamin D insufficiency was associated with severity of oxidative stress in vitiligo patients. Our work also provides new insights into the mechanism of vitamin D against vitiligo, in which vitamin D protects melanocytes against oxidative stress by activating Wnt/β-catenin signaling.
Similar content being viewed by others


Introduction
Vitiligo is an acquired skin disease characterized by progressive depigmentation due to localized destruction of epidermal melanocytes. The etiology of vitiligo is still not fully understood, but several factors have been implicated in the melanocyte destruction, such as oxidative stress, autoimmunity, and genetic predisposition [1,2,3,4]. Among them, oxidative stress has been attracting increasing attention. It can directly damage lipids and proteins, leading to functional impairment or cell apoptosis of melanocytes, and also initiate the autoimmune response that subsequently mediates melanocyte cytotoxicity 5, 6]. Therefore, antioxidant treatment has become a promising therapeutic direction for vitiligo.
Vitamin D exerts potent physiological and pharmacological actions through its nuclear receptor (VDR) in various cell types, and thus it has great potential for treating a variety of diseases such as osteoporosis, cancer, and autoimmune diseases [7]. Vitamin D analogs (particularly calcipotriol and tacalcitol) have been widely utilized as topical therapeutic agents in vitiligo [8, 9]. In addition, serum vitamin D insufficiency and VDR gene polymorphisms were also reported in vitiligo patients, further highlighting the significance of vitamin D against vitiligo [10,11,12]. The multipronged effect of vitamin D analogs against vitiligo has been confirmed in several studies. For example, vitamin D compounds can modulate the proliferation, differentiation, migration, and apoptosis of melanocytes, and influence the local immune response by regulating T cell activation in vitiligo [8, 13,14,15]. A recently proposed mechanism involving the pharmacological action of vitamin D against vitiligo is based on its antioxidant properties since oxidative stress is crucial during the onset and progression of vitiligo [16, 17]. However, the molecular mechanism by which vitamin D protects human melanocytes against oxidative damage is still not clear.
Canonical Wnt signaling is a fundamental mechanism that governs cell proliferation, differentiation, and apoptosis by regulating the amount of the transcriptional co-activator β-catenin [18,19,20]. In the absence of Wnt signaling, cytoplasmic β-catenin is constitutively degraded via destruction of the Axin complex. When Wnt binds to Frizzled receptor (Fz) and its co-receptor (LRP6 or LRP5), Wnt/β-catenin signaling is activated. Formation of the Wnt–Fz–LRP6 complex together with recruitment of the scaffolding protein Dishevelled (Dvl) further mediates the disassembly of the destruction complex, and thus the stabilization of β-catenin, which accumulates and travels to the nucleus and activates target gene expression. β-catenin stabilization is negatively associated with the activity of glycogen synthase kinase 3β (GSK3β), which is an essential component of the destruction complex [20]. Recently, some studies have found that Wnt/β-catenin signaling is inhibited in H2O2-treated keratinocytes and melanocytes, while Wnt agonists trigger melanocyte differentiation and melanogenesis in vitiligo [21, 22]. In addition, Wnt/β-catenin pathway has also been shown to be responsible for the protection of vitamin D against oxidative stress in other types of cells such as osteoblasts and nephrocytes [23, 24]. Thus, we hypothesized that vitamin D protects human melanocytes from oxidative stress by activating Wnt/β-catenin signaling.
Materials and methods
Patient and control specimens
After obtaining informed written consent, blood samples were collected from 53 vitiligo patients with non-segmental disease (29 men and 24 women; 37 active and 16 stable; age range, 11–57 years; mean age, 30 years) and from 23 healthy controls (10 men and 13 women; age range, 18–60 years; mean age, 33 years). The lesional area in all vitiligo patients was more than 3% of the body area. None of the patients underwent any systemic or topical treatment 3 months before the blood samples were obtained. This study was approved by the local ethics committee of Huashan Hospital and performed in strict compliance with the principles of the Declaration of Helsinki.
Serum MDA and vitamin D measurement
Serum MDA levels were tested using the thiobarbituric acid (TBA) reactive substances assay (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) [25]. MDA is an oxidative degradation product of lipids, which reacts with TBA and then forms red complexes with the absorbent at 532 nm. Serum vitamin D (25-hydroxyvitamin D) was detected using an enzyme-linked immunosorbent assay kit (Groundwork Biotechnology Diagnosticate Ltd, San Diego, CA, USA), according to the manufacturer’s protocol. The 25-hydroxyvitamin D levels were divided into ‘sufficiency’ (>75 nmol/L), ‘insufficiency’ (≥25 nmol/L but <75 nmol/L), and ‘deficiency’ (<25 nmol/L) as previously reported [26].
Cell culture and treatment
The immortalized human epidermal melanocyte cell lines PIG1 (normal melanocytes) and PIG3V (vitiligo melanocytes) were gifts from Dr. Caroline Le Poole (Loyola University Chicago, Maywood, IL) and cultured in Medium 254 (Cascade Biologics/Invitrogen, Portland, OR, USA) supplemented with human melanocyte growth supplement (Cascade Biologics/Invitrogen), 5% fetal bovine serum (Invitrogen, CA, USA), and penicillin–streptomycin antibiotic mix (Invitrogen) at 37 °C in the presence of 5% CO2 [27]. Melanocytes were pretreated with 1 nM 1,25-dihydroxyvitamin D3 (D1530, Sigma) or 40 μM SKL2001 (S8230, Selleck, USA) for 24 h as in previous studies [21, 28]. Oxidative stress was then induced by addition of 0.5 mM H2O2 (Sigma) for another 24 h.
Determination of cell viability by the CCK-8 assay
Cell viability was measured using the CCK-8 assay (Cell Counting Kit-8; Beyotime Institute of Biotechnology, Haimen, China) to count living cells following the manufacturer’s protocol. PIG1 or PIG3V cells were seeded into 96-well plates (3 × 103 cells per well) in quadruplicate. After treatment with the indicated agents, 10 μL of CCK-8 reagent was added to 90 μL medium and incubated for 90 min at 37 °C. Cell viability was determined by monitoring the color change using an ELISA plate reader (Bio-Rad, Hercules, CA, USA) at an absorbance of 450 nm.
Apoptosis detection by Annexin V-FITC
Melanocytes were cultured in six-well plates at a density of 1.2 × 106 cells for PIG1 and 0.8 × 106 cells for PIG3V. Cell apoptosis was evaluated using the Annexin V-FITC Apoptosis Detection Kit (MaiBio, Shanghai, China). FITC fluorescence was detected using a flow cytometer (Beckman Coulter, Miami, USA) and analyzed with the Expo32 software (Beckman Coulter).
Transwell migration assay
Cell migration was evaluated in Transwell cell culture chambers (Costar 3422; Cambridge, MA, USA) as previously described [29]. PIG1 and PIG3V cells were treated with 0.5 mM H2O2 for 24 h and then seeded into the top chambers of the Transwell in medium containing 1 nM 1,25-dihydroxyvitamin D3 or 40 μM SKL2001 for 48 h. The bottom chambers of the Transwell were filled with 500 μL 10% FBS supplemented with different treatments. Non-migrating cells on the top surface of the membrane were scraped with a cotton swab, and the cells that had traveled to the bottom side (migrating cells) were fixed with cold 4% paraformaldehyde for 10 min. Migrating cells were stained with 0.1% hexamethyl pararosaniline, imaged with a Nikon microscope, and then quantified by manual counting.
Measurement of intracellular reactive oxygen species (ROS)
Melanocytes were seeded into 96-well plates at a density of 6 × 103 cells for PIG1 and 3 × 103 cells for PIG3V. Following the indicated treatments, intracellular ROS levels were determined using the fluorescent probe H2DCFDA (Thermofisher, MA, USA) according to the manufacturer’s protocol.
RNA isolation and quantitative real-time PCR
Total RNA was isolated using TRIzol reagent (Invitrogen) and then reverse-transcribed into cDNA using the SuperScript III First-Strand Synthesis Kit (Invitrogen). Subsequent real-time PCR was performed using iQ SYBR Green Supermix (Bio-Rad) per the manufacturer’s recommendations. Primers (Supplementary Material, Table S1) were synthesized by Shanghai Sangon Biotech Co., Ltd (Shanghai, China).
Western blot analysis
Nuclear-cytoplasmic fractionation was carried out using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, USA). Proteins were extracted from cells after the indicated treatment and quantified using the BCA protein assay kit (Pierce, Rockford, IL, USA). Equal amounts of proteins were loaded into 8% SDS-PAGE and then transferred onto a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). After blocking and washing, the membranes were incubated with the indicated primary Abs and then with horseradish peroxidase-labeled secondary antibody. The blots were assessed using ImageJ software (NIH, Bethesda, MD). All antibodies (Abs) and their sources are listed in Table S2.
RNA interference
The siRNA for beta-catenin (Supplementary Material, Table S1) was purchased from Genepharm technology, China. Melanocyte cells were seeded at 2 × 105 cells per well for 24 h before transfection. Cells were transfected with beta-catenin siRNA and an irrelevant siRNA control at 50 nM (GenePharma, China) with LipofectamineTM 2000 (Invitrogen, Carlsbad, USA) following the manufacturer’s protocol.
Statistical analyses
The data represent the mean ± SD of at least three independent experiments. All statistical analyses were performed using the GraphPad Prism version 5.0 software (GraphPad Software, San Diego, CA, USA). Dual comparisons were carried out with the two-tailed Student’s unpaired t test. Groups of three or more were analyzed by one-way analysis of variance (ANOVA) with Dunnett’s post-tests. The Pearson correlation coefficient was calculated to examine the association between serum MDA and vitamin D levels. P values < 0.05 were considered statistically significant.
Results
Decreased serum vitamin D levels correlated with increased serum MDA in vitiligo patients
Malondialdehyde (MDA) is a specific indicator of lipid peroxidation, reflecting the extent of oxidative damage in vitiligo patients. To investigate the possible role of Vitamin D in the pathogenesis of vitiligo, serum levels of vitamin D and MDA were first tested in 53 patients with non-segmental vitiligo (lesional area ≥ 3%) and 23 healthy control subjects by using ELISA. Median serum MDA levels were significantly higher in the patients with vitiligo (8.02 ± 3.22 nmol/mL) than in healthy controls (6.54 ± 1.68 nmol/mL) (Fig. 1a). However, the overall level of serum vitamin D in vitiligo patients (34.23 ± 13.57 nmol/L) was clearly lower than that in healthy controls (45.80 ± 12.01 nmol/L; Fig. 1b). The rate of vitamin D deficiency was 24.5% in vitiligo patients as compared with none in healthy controls. Moreover, we also assessed the relationship between serum vitamin D levels and MDA levels in vitiligo patients. Statistical analysis suggested that the level of serum vitamin D was inversely correlated with that of serum MDA in vitiligo patients (r = −0.3185, P = 0.0201, n = 53; Fig. 1c).

Serum vitamin D insufficiency or decreased VDR expression was associated with vitiligo pathogenesis. a Decreased serum vitamin D levels and (b) increased serum MDA levels in patients with vitiligo. Ctrl healthy controls, PV progressive vitiligo, SV stable vitiligo, Total total patients. c Association between serum vitamin D and MDA levels. Serum vitamin D levels were inversely correlated with serum MDA levels in vitiligo patients (r = −0.3185, p = 0.0201, n = 53, Spearman’s correlation test). The solid line represents the regression line. d Vitiligo melanocytes showed lower expression of vitamin D receptor (VDR) than normal melanocytes. *P < 0.05, **P < 0.01, ***P < 0.001
Since vitamin D exerted pleiotropic biological effects on the growth and differentiation of cutaneous cells via the nuclear vitamin D receptor (VDR) [30], we further assessed the expression of VDR in both normal (PIG1) and vitiligo melanocytes (PIG3V). Protein blotting showed that the protein level of VDR was significantly lower in PIG3V than PIG1 (P < 0.001; Fig. 1d). Taken together, these results indicated that serum vitamin D insufficiency and functional impairment was involved in vitiligo pathogenesis, which might be associated with oxidative damage in melanocytes.
Vitamin D activated the WNT/β-catenin pathway in human melanocytes under oxidative stress
Since the β-catenin pathway was involved in the protection of vitamin D against various skin diseases [30, 31], we next evaluated the effect of vitamin D on the expression of β-catenin pathway components in both normal and vitiligo melanocytes. A concentration of 1 nM vitamin D (1,25-dihydroxyvitamin D3) was selected for dosing according to our previous study, using the Wnt agonist SKL2001 (40 μM) as a positive control [21, 28, 32]. Melanocytes were pretreated with vitamin D or SKL2001 and then challenged with 0.5 mM H2O2 for 24 h. As shown in Fig. 2, 0.5 mM H2O2 significantly downregulated the expression of β-catenin and its downstream effector CDH3 at both mRNA and protein levels in both PIG1 and PIG3V cells. Vitamin D partially reversed the inhibitory effect of oxidative stress on the expression of β-catenin in both H2O2-treated PIG1 and PIG3V, similarly to SKL2001 (Fig. 2a-d). We further showed that vitamin D significantly enhanced the nuclear/cytosolic ratio of β-catenin in these two melanocytes with or without oxidative stress. Consistently, vitamin D also induced a significant enhancement of CDH3 expression at both mRNA and protein levels in both PIG1 and PIG3V under oxidative stress, demonstrating the functional activation of β-catenin. Therefore, vitamin D might promote nuclear translocation and transcriptional activity of β-catenin in human melanocytes under oxidative stress.

Vitamin D promoted the activation of Wnt/β-catenin signaling in H2O2-treated melanocytes. Melanocytes were exposed to 1 nM vitamin D or 40 μM SKL2001 for 24 h and then further treated with 0.5 mmol/L H2O2 for another 24 h. β-catenin and CDH3 mRNA levels were analyzed by quantitative real-time PCR in PIG1 (a) and PIG3V (b). c Western blots of nuclear and cytoplasmic fractions of β-catenin and total CDH3. d The ratio of nuclear/cytosolic of β-catenin was calculated by densitometry analysis. (e) Western blots of total GSK3β and phosphorylated GSK3β at Ser9. f The ratio of p-GSK3β (Ser9)/GSK3β was analyzed by densitometry analysis. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the H2O2 group
GSK3β is an important negative regulator of β-catenin signaling, and the stabilization of β-catenin is directly proportional to GSK3β phosphorylation at Ser9 [18, 20]. As shown in Fig. 2e, f, vitamin D markedly increased the ratio of p-GSK3β (Ser9)/GSK3β in melanocytes even without H2O2 treatment, indicating that vitamin D might induce GSK3β phosphorylation on Ser9 and thus promote GSK3β inactivation and nuclear translocation of β-catenin. Taken together, vitamin D activated WNT/β-catenin signaling in both normal and vitiligo melanocytes against oxidative stress.
Vitamin D ameliorates H2O2-induced oxidative damage in human melanocytes similar to Wnt agonist
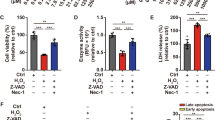
To further assess the protective effects of vitamin D against vitiligo, a series of in vitro assays were performed in both normal and vitiligo melanocytes. As shown in Fig. 3a, H2O2 treatment significantly increased intracellular ROS production in human melanocytes, especially PIG3V. Nevertheless, pretreatment with vitamin D or SKL2001 attenuated H2O2-induced ROS accumulation. Moreover, CCK8 and Transwell assays further revealed that either vitamin D or SKL2001 dramatically increased the cell viability and migratory rates of H2O2-treated melanocytes (Fig. 3b, c, Fig. S1). Similarly to previous studies, we also found that H2O2 exposure increased the apoptotic rates of PIG1 and PIG3V approximately five- to six-fold, whereas pretreatment with vitamin D or SKL2001 markedly reduced melanocytes apoptosis by 66–70% (Fig. 3d, Fig. S2). Although vitiligo melanocytes were more susceptible to oxidative stress than normal ones, vitamin D displayed potent protective effects on the proliferation, migration, and apoptosis of PIG3V similarly to those on PIG1. Taken together, these results indicated that vitamin D could reverse H2O2-induced oxidative damage of human melanocytes similar to the WNT agonist.

Vitamin D attenuated H2O2-induced oxidative damage in human melanocytes similarly to Wnt agonist. a Measurement of intracellular ROS. b Cell viability determination by the CCK-8 assay. c Cell migration assay. d Cell apoptosis assay. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the H2O2 group
β-catenin silencing abolished the protective effect of Vitamin D on melanocytes against oxidative stress
To test whether the WNT/β-catenin pathway is required for the effects of vitamin D protecting melanocytes against oxidative stress, we used a specific short interfering RNA (siRNA) to downregulate the expression of β-catenin. Quantitative real-time PCR and immunoblotting showed significant changes in β-catenin mRNA and protein levels after β-catenin siRNA#2 (S2) transfection compared with scrambled siRNA (NC) (Fig. 4a, b). In subsequent experiments, PIG1 and PIG3V cells were pretreated with NC or S2 for phenotypic analyses. β-catenin deficiency markedly abrogated the downregulation of ROS production in vitamin D-treated melanocytes under oxidative stress (Fig. 4c). In addition, vitamin D induced enhancement of the proliferation and migration of H2O2-treated melanocytes was also abolished by β-catenin silencing (Fig. 4d, e, Fig. S3). Consistently, β-catenin silencing also neutralized the protection of vitamin D against H2O2-induced apoptosis (Fig. 4f, Fig. S4). Both PIG1 and PIG3V cells with β-catenin silencing displayed similar phenotypes in vitro under oxidative stress. These results suggested that β-catenin activation was essential for the protective capacity of vitamin D in melanocytes against oxidative stress.

β-catenin silencing abolished the protection of vitamin D in H2O2-treated melanocytes. Quantitative real-time PCR (a) and Western blotting (b) were carried out to evaluate β-catenin expression in melanocytes transfected with β-catenin siRNA and an irrelevant siRNA control. BC blank control, NC negative control, S1-S3 β-catenin siRNA#1-#3. c Measurement of intracellular ROS. d Cell viability determination by the CCK-8 assay. e Cell migration assay. f Cell apoptosis assay. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the H2O2 group
Vitamin D modulated Nrf2, MITF, and apoptosis pathways by activation of β-catenin in melanocytes
Previous studies have shown that Wnt/β-catenin signaling controls cell proliferation, differentiation, and apoptosis by regulating a large number of target genes [19, 33]. We first examined the alterations of the Nrf2/ARE pathway, which is a master regulator that governs the redox balance of melanocytes [27, 34]. We found that H2O2 exposure induced a significant decrease in the ratio of nuclear/cytosolic of Nrf2 and the expression of HO-1 in normal melanocytes (Fig. 5a-c). Impairment of the Nrf2/ARE pathway caused by H2O2 was markedly reversed by addition of vitamin D, while activation of the Nrf2/ARE pathway induced by vitamin D was further blocked by β-catenin silencing. Therefore, β-catenin pathways mediated the activation of Nrf2 in vitamin D-treated melanocytes against oxidative damage.

β-catenin activation was required for vitamin D to modulate Nrf2, MITF, and apoptosis pathways in melanocytes. a Western blotting was performed to assess the expression of downstream components in Wnt/β-catenin signaling. The ratio of nuclear/cytosolic of Nrf2 (b), total HO-1 and MITF (c), and the ratio of cleaved-caspase3/total caspase 3 (d) were analyzed by densitometry analysis. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the H2O2 group
Microphthalmia-associated transcription factor (MITF) is an essential transcription factor involved in melanocyte differentiation, migration, and melanogenesis. We found that vitamin D treatment significantly upregulated the protein level of MITF in PIG1 cells with or without β-catenin silencing (Fig. 5a, c). Moreover, vitamin D also notably reversed the inhibitory effect of H2O2 on MITF expression, but the protection of vitamin D was abolished by β-catenin deficiency. The results indicated that vitamin D might positively modulate MITF to promote melanocyte proliferation and migration by activating the β-catenin pathway.
In addition to Nrf2 and MITF, the apoptotic pathway is another important target of the WNT/β-catenin pathway [33]. Thus, expression of caspase 3 and its cleaved form were tested in melanocytes. Consistent with the increased apoptotic rates of melanocytes, H2O2 exposure induced a ten-fold upregulation of the ratio of cleaved-caspase3/caspase3 (Fig. 5a, d). Vitamin D treatment remarkably clearly inhibited the upregulation of cleaved caspase 3, while its protective role was further blocked by β-catenin silencing. These results suggested that the β-catenin pathway was required for the anti-apoptotic effect of vitamin D on human melanocytes.
Discussion
Recent evidence indicates that treatment with vitamin D analogs enhances repigmentation in vitiligo, especially in combination with UV light or corticosteroids [8]. However, the molecular mechanisms underlying the pharmacological action of vitamin D compounds against vitiligo have not yet been investigated. In the present study, we revealed a new mechanism of vitamin D as an antioxidative agent for vitiligo treatment. We found that vitamin D protected melanocytes against oxidative damage by activating the WNT/β-catenin pathway. The β-catenin pathway was required for vitamin D to modulate downstream targets such as Nrf2/ARE, MITF, and apoptotic pathways.
Oxidative stress is a critical factor in the onset and progression of vitiligo [1, 2, 6, 27]. Consistent with previous studies [10, 35,36,37], we found that patients with vitiligo had higher MDA levels and lower vitamin D levels than the healthy control group. Intriguingly, the decreased serum vitamin D level was inversely correlated with serum MDA levels in vitiligo patients, indicative of the potential antioxidant effect of vitamin D. A series of in vitro experiments was conducted and further confirmed that vitamin D attenuated H2O2-induced ROS accumulation in human melanocytes and ameliorated the effect of oxidative stress on proliferation, migration, and apoptosis of melanocytes. Furthermore, vitiligo melanocytes displayed a significant decrease in protein levels of VDR compared with normal melanocytes. Our results were highly consistent with previous studies which revealed that VDR expression was remarkably decreased in the lesional/perilesional skin while repigmentation was accompanied with upregulated VDR expression in vitiligo patients [38, 39]. The VDR gene polymorphisms were also reported to be associated with serum 25-hydroxyvitamin D levels in Chinese patients with generalized vitiligo [12]. Therefore, vitamin D insufficiency or functional impairment of the vitamin D pathway might attenuate the antioxidant capacity of patients and thus represent a risk factor for vitiligo.
It has been reported that vitiligo patients with comorbid autoimmune illness have lower levels of 25-hydroxyvitamin D [11]. The mechanism by which vitamin D modulates autoimmunity is unknown, which might be explained by the following possible reasons. On the one hand, vitamin D has multiple effects on innate and adaptive immune responses by regulating various immune cells in vitro [40]. On the other hand, oxidative stress has been shown to be an important initiate factor in the pathogenesis of various autoimmune diseases [6, 41, 42], and vitamin D ameliorates oxidative damage and thus reduces the incidence and severity of autoimmune responses. The antioxidant property of vitamin D has been validated in various human cells, such as cardiac cells, endothelial cells, gastric epithelial cells, and melanocytes [16, 43,44,45]. In the present study, we further showed that vitamin D could protect melanocytes against oxidative damage by activating the canonical WNT/β-catenin pathway.
A previous study by Regazzetti et al has demonstrated an inhibition of the WNT/β-catenin pathway in keratinocytes and melanocytes under oxidative stress [21]. Moreover, lesional vitiligo skin is also characterized by downregulated expression of WNT pathway components such as LEF1, CDH2, CDH3. Similarly, in our study, vitiligo melanocytes displayed lower expression of CDH3 (p-cadherin) and nuclear β-catenin than normal melanocytes. Because β-catenin is a transcription factor, nuclear translocation of β-catenin is critical for its dissociation from GSK3β and transcriptional activation of multiple target genes [18, 33]. We further found that in addition to downregulation of CDH3 and β-catenin expression, H2O2 also decreased the nuclear/cytosolic ratio of β-catenin in both PIG1 and PIG3V and prevented GSK3β phosphorylation on Ser9 in PIG3V, indicating that oxidative stress inhibited the nuclear translocation of β-catenin. The inhibitory effects on β-catenin signaling induced by H2O2 were reversed by addition of vitamin D or WNT agonist (SKL2001). Thus, vitamin D positively modulated β-catenin signaling at both translational and posttranslational levels in melanocytes under oxidative stress.
A previous study has shown that WNT agonist induces the differentiation of resident stem cells into pre-melanocytes in vitiligo lesional skin ex vivo [21]. Our study further highlighted that WNT pathway activation was essential for the antioxidant properties of vitamin D in melanocytes. Like the WNT agonist, vitamin D significantly inhibited ROS accumulation and cell apoptosis in H2O2-treated melanocytes, and promoted their proliferative and migratory activity. The protective effects of vitamin D against oxidative stress were abolished by β-catenin silencing in melanocytes. Similar protective effects and mechanisms of vitamin D have been reported in other cells and tissues [23, 24, 46, 47]. For example, activated β-catenin signaling is required for vitamin D to promote osteogenesis in oxidative stress, while inhibition of the WNT pathway is involved in the protection of vitamin D against colorectal tumorigenesis [23, 47]. Thus, vitamin D might utilize different modulatory patterns of the WNT/β-catenin pathway to protect against various diseases.
The transcription factor Nrf2 is a critical regulator of oxidant metabolism in melanocytes, and it is required for the effectiveness of aspirin and simvastatin as novel therapeutic strategies against vitiligo [27, 48,49,50]. Several studies also revealed that Nrf2 signaling is involved in the protective effects of vitamin D in various disease models [51,52,53]. Consistently, our results also showed that vitamin D promoted the upregulation of nuclear Nrf2 and its downstream component HO-1 in melanocytes. Furthermore, activation of Nrf2 signaling was completely blocked by β-catenin silencing, indicating that the β-catenin pathway mediated Nrf2 activation in vitamin D-treated melanocytes. Recently, a number of studies have explored the relationship between the Wnt/β-catenin and Nrf2 pathways [54,55,56,57]. For example, WNT-3A controls the antioxidant metabolism of hepatocytes by activating Nrf2 in a β-catenin-independent manner, and Nrf2 pathway disruption induces neurogenic impairment in Parkinsonian mice via Wnt/β-catenin dysregulation [54, 57]. Therefore, Wnt/β-catenin signaling has complex interactions with Nrf2 signaling that might be specific to the cell type and disease model. The potential mechanism by which β-catenin activates Nrf2 signaling in melanocytes remains to be further investigated.
In addition to Nrf2 signaling, we also found that MITF and apoptotic pathways were regulated by β-catenin in vitamin D-treated melanocytes. MITF is a central regulator of the differentiation, migration, survival, and melanogenesis of melanocytes [58,59,60]. Previous studies have shown that MITF is significantly upregulated by WNT/β-catenin pathway activation in both vitiligo lesional skin ex vivo and hair follicle melanocytes in vitro [21, 61]. Consistently, we observed that vitamin D treatment enhanced the expression of MITF in human melanocytes. In our study, however, β-catenin deficiency abolished the upregulation of MITF in melanocytes under oxidative stress, but not in melanocytes without H2O2 exposure. A previous study by Watabe et al. once showed that vitamin D induced MITF expression by stimulating endothelin B receptor signaling [13]. Thus, we speculated that vitamin D could modulate MITF through multiple pathways and Wnt/β-catenin signaling might be crucial for MITF regulation in melanocytes under oxidative stress. These hypotheses remain to be validated in further experiments. We also showed that β-catenin activation was essential for vitamin D to modulate apoptotic pathway in melanocytes. When β-catenin was silenced, vitamin D could not reverse the increase in cleaved caspase 3 expression or apoptotic rates in H2O2-treated melanocytes.
In conclusion, our study shows that vitamin D protects human melanocytes against oxidative damage by activation of Wnt/β-catenin signaling, indicating targeting Wnt/β-catenin signaling could be a useful approach to improve treatment of vitiligo. Serum vitamin D insufficiency or functional impairment of the vitamin D pathway might be a risk factor for vitiligo due to impairment of the antioxidant system in vivo. Further studies using vitiligo mice are necessary to confirm the therapeutic potential of systemic administration of vitamin D for vitiligo.
References
Colucci R, Dragoni F, Moretti S. Oxidative stress and immune system in vitiligo and thyroid diseases. Oxid Med Cell Longev. 2015;2015:631927.
Denat L, Kadekaro AL, Marrot L, et al. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol. 2014;134:1512–8.
Speeckaert R, Speeckaert MM, van Geel N. Why treatments do(n’t) work in vitiligo: An autoinflammatory perspective. Autoimmun Rev. 2015;14:332–40.
Jin Y, Birlea SA, Fain PR, et al. Genome-wide analysis identifies a quantitative trait locus in the MHC class II region associated with generalized vitiligo age of onset. J Invest Dermatol. 2011;131:1308–12.
Dell’Anna ML, Ottaviani M, Albanesi V, et al. Membrane lipid alterations as a possible basis for melanocyte degeneration in vitiligo. J Invest Dermatol. 2007;127:1226–33.
Li S, Zhu G, Yang Y, et al. Oxidative stress drives CD8+ T-cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J Allergy Clin Immunol. 2017;140(1):177–189.
Sintov AC, Yarmolinsky L, Dahan A, et al. Pharmacological effects of vitamin D and its analogs: recent developments. Drug Discov Today. 2014;19:1769–74.
Birlea SA, Costin GE, Norris DA. New insights on therapy with vitamin D analogs targeting the intracellular pathways that control repigmentation in human vitiligo. Med Res Rev. 2009;29:514–46.
Parsad D, Kanwar AJ. Topical vitamin D analogues in the treatment of vitiligo. Pigment Cell Melanoma Res. 2009;22:487–8.
Upala S, Sanguankeo A. Low 25-hydroxyvitamin D levels are associated with vitiligo: a systematic review and meta-analysis. Photodermatol Photoimmunol Photomed. 2016;32:181–90.
Silverberg JI, Silverberg AI, Malka E, et al. A pilot study assessing the role of 25 hydroxy vitamin D levels in patients with vitiligo vulgaris. J Am Acad Dermatol. 2010;62:937–41.
Li K, Shi Q, Yang L, et al. The association of vitamin D receptor gene polymorphisms and serum 25-hydroxyvitamin D levels with generalized vitiligo. Br J Dermatol. 2012;167:815–21.
Watabe H, Soma Y, Kawa Y, et al. Differentiation of murine melanocyte precursors induced by 1,25-dihydroxyvitamin D3 is associated with the stimulation of endothelin B receptor expression. J Invest Dermatol. 2002;119:583–9.
Birlea SA, Costin GE, Norris DA. Cellular and molecular mechanisms involved in the action of vitamin D analogs targeting vitiligo depigmentation. Curr Drug Targets. 2008;9:345–59.
Kawakami T, Ohgushi A, Hirobe T, et al. Effects of 1,25-dihydroxyvitamin D3 on human epidermal melanocytes and melanoblasts. J Dermatol Sci. 2014;76:72–4.
Gong Q, Li X, Sun J, et al. The effects of calcipotriol on the dendritic morphology of human melanocytes under oxidative stress and a possible mechanism: is it a mitochondrial protector? J Dermatol Sci. 2015;77:117–24.
Li QL, Wu YH, Niu M, et al. Protective effects of tacalcitol against oxidative damage in human epidermal melanocytes. Int J Dermatol. 2017;56:232–8.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26.
Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810.
Kimelman D, Xu W. beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–91.
Regazzetti C, Joly F, Marty C, et al. Transcriptional analysis of vitiligo skin reveals the alteration of WNT pathway: a promising target for repigmenting vitiligo patients. J Invest Dermatol. 2015;135:3105–14.
Yamada T, Hasegawa S, Inoue Y, et al. Wnt/beta-catenin and kit signaling sequentially regulate melanocyte stem cell differentiation in UVB-induced epidermal pigmentation. J Invest Dermatol. 2013;133:2753–62.
Xiong Y, Zhang Y, Xin N, et al. 1alpha,25-Dihydroxyvitamin D3 promotes osteogenesis by promoting Wnt signaling pathway. J Steroid Biochem Mol Biol. 2017;174:153–60.
Guo J, Lu C, Zhang F, et al. VDR activation reduces proteinuria and high-glucose-induced injury of kidneys and podocytes by regulating wnt signaling pathway. Cell Physiol Biochem. 2017;43:39–51.
Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–8.
Xu X, Fu WW, Wu WY. Serum 25-hydroxyvitamin D deficiency in Chinese patients with vitiligo: a case-control study. PLoS ONE. 2012;7:e52778.
Jian Z, Li K, Song P, et al. Impaired activation of the Nrf2-ARE signaling pathway undermines H2O2-induced oxidative stress response: a possible mechanism for melanocyte degeneration in vitiligo. J Invest Dermatol. 2014;134:2221–30.
Tang Luyan, Fu Wenwen, Zhang Yong, et al. Effect of calcipotriol on melanin synthesis of human melanocytes. Chin J Dermatol. 2009;42:771–3.
Lei TC, Vieira WD, Hearing VJ. In vitro migration of melanoblasts requires matrix metalloproteinase-2: implications to vitiligo therapy by photochemotherapy. Pigment Cell Res. 2002;15:426–32.
Slominski AT, Janjetovic Z, Kim TK, et al. Novel non-calcemic secosteroids that are produced by human epidermal keratinocytes protect against solar radiation. J Steroid Biochem Mol Biol. 2015;148:52–63.
Shah S, Islam MN, Dakshanamurthy S, et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Mol Cell. 2006;21:799–809.
Gwak J, Hwang SG, Park HS, et al. Small molecule-based disruption of the Axin/beta-catenin protein complex regulates mesenchymal stem cell differentiation. Cell Res. 2012;22:237–47.
Vlad A, Rohrs S, Klein-Hitpass L, et al. The first five years of the Wnt targetome. Cell Signal. 2008;20:795–802.
Jian Z, Li K, Liu L, et al. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J Invest Dermatol. 2011;131:1420–7.
Ines D, Sonia B, Riadh BM, et al. A comparative study of oxidant-antioxidant status in stable and active vitiligo patients. Arch Dermatol Res. 2006;298:147–52.
Khan R, Satyam A, Gupta S, et al. Circulatory levels of antioxidants and lipid peroxidation in Indian patients with generalized and localized vitiligo. Arch Dermatol Res. 2009;301:731–7.
Saleh HM, Abdel Fattah NS, Hamza HT. Evaluation of serum 25-hydroxyvitamin D levels in vitiligo patients with and without autoimmune diseases. Photodermatol Photoimmunol Photomed. 2013;29:34–40.
Doss RW, El-Rifaie AA, Gohary YM, et al. Vitamin D receptor expression in vitiligo. Indian J Dermatol. 2015;60:544–8.
El-Hanbuli HM, Dawoud NM, Mahmoud RH. Narrow-band UVB effects on cutaneous vitamin D receptor expression and serum 25-hydroxyvitamin D in generalized vitiligo. Photodermatol Photoimmunol Photomed. 2018;34:175–83.
Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4:404–12.
Adamczyk B, Adamczyk-Sowa M. New Insights into the role of oxidative stress mechanisms in the pathophysiology and treatment of multiple sclerosis. Oxid Med Cell Longev. 2016;2016:1973834.
Harris JE. Cellular stress and innate inflammation in organ-specific autoimmunity: lessons learned from vitiligo. Immunol Rev. 2016;269:11–25.
Farhangi MA, Nameni G, Hajiluian G, et al. Cardiac tissue oxidative stress and inflammation after vitamin D administrations in high fat- diet induced obese rats. BMC Cardiovasc Disord. 2017;17:161.
Haas MJ, Jafri M, Wehmeier KR, et al. Inhibition of endoplasmic reticulum stress and oxidative stress by vitamin D in endothelial cells. Free Radic Biol Med. 2016;99:1–10.
Uberti F, Bardelli C, Morsanuto V, et al. Role of vitamin D3 combined to alginates in preventing acid and oxidative injury in cultured gastric epithelial cells. BMC Gastroenterol. 2016;16:127.
Lee H, Bae S, Yoon Y. Anti-adipogenic effects of 1,25-dihydroxyvitamin D3 are mediated by the maintenance of the wingless-type MMTV integration site/beta-catenin pathway. Int J Mol Med. 2012;30:1219–24.
Groschel C, Aggarwal A, Tennakoon S, et al. Effect of 1,25-dihydroxyvitamin D3 on the Wnt pathway in non-malignant colonic cells. J Steroid Biochem Mol Biol. 2016;155:224–30. (Pt B)
Natarajan VT, Singh A, Kumar AA, et al. Transcriptional upregulation of Nrf2-dependent phase II detoxification genes in the involved epidermis of vitiligo vulgaris. J Invest Dermatol. 2010;130:2781–9.
Jian Z, Tang L, Yi X, et al. Aspirin induces Nrf2-mediated transcriptional activation of haem oxygenase-1 in protection of human melanocytes from H2O2 -induced oxidative stress. J Cell Mol Med. 2016;20:1307–18.
Chang Y, Li S, Guo W, et al. Simvastatin protects human melanocytes from H2O2-induced oxidative stress by activating Nrf2. J Invest Dermatol. 2017;137:1286–96.
Wang Z, Zhang H, Sun X, et al. The protective role of vitamin D3 in a murine model of asthma via the suppression of TGF-beta/Smad signaling and activation of the Nrf2/HO-1 pathway. Mol Med Rep. 2016;14:2389–96.
Nakai K, Fujii H, Kono K, et al. Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am J Hypertens. 2014;27:586–95.
Teixeira TM, da Costa DC, Resende AC, et al. Activation of Nrf2-antioxidant signaling by 1,25-dihydroxycholecalciferol prevents leptin-induced oxidative stress and inflammation in human endothelial cells. J Nutr. 2017;147:506–13.
Rada P, Rojo AI, Offergeld A, et al. WNT-3A regulates an Axin1/NRF2 complex that regulates antioxidant metabolism in hepatocytes. Antioxid Redox Signal. 2015;22:555–71.
Long MJ, Lin HY, Parvez S, et al. beta-TrCP1 Is a vacillatory regulator of Wnt signaling. Cell Chem Biol. 2017;24:944–957.e947.
Manigandan K, Manimaran D, Jayaraj RL, et al. Taxifolin curbs NF-kappaB-mediated Wnt/beta-catenin signaling via up-regulating Nrf2 pathway in experimental colon carcinogenesis. Biochimie. 2015;119:103–12.
L’Episcopo F, Tirolo C, Testa N, et al. Aging-induced Nrf2-ARE pathway disruption in the subventricular zone drives neurogenic impairment in parkinsonian mice via PI3K-Wnt/beta-catenin dysregulation. J Neurosci. 2013;33:1462–85.
Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–14.
Cheli Y, Ohanna M, Ballotti R, et al. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010;23:27–40.
Seberg HE, Van Otterloo E, Cornell RA. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res. 2017;30:454–66.
Guo H, Yang K, Deng F, et al. Wnt3a promotes melanin synthesis of mouse hair follicle melanocytes. Biochem Biophys Res Commun. 2012;420:799–804.
Acknowledgements
This study was supported by grants from Municipal Hospital Emerging Frontier Technology United Key Projects (SHDC12016112), the Shanghai Key Medical Discipline for Dermatology (2017ZZ02002), Shanghai leading talent project, and China Postdoctoral Science Foundation (2016M600286). The authors thank Dr. Caroline Le Poole (Loyola University Chicago, Maywood, IL, US) for providing the human melanocyte cell lines PIG1 and PIG3V and Dr. Chunying Li (Xijing Hospital, Fourth Military Medical University, Xi’an, Shaanxi, China) for assisting with the cell transfer. The authors also thank the vitiligo patients and healthy donors for their participation in this study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no financial conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Tang, L., Fang, W., Lin, J. et al. Vitamin D protects human melanocytes against oxidative damage by activation of Wnt/β-catenin signaling. Lab Invest 98, 1527–1537 (2018). https://doi.org/10.1038/s41374-018-0126-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0126-4
