
Abstract
Since the discovery of the lysosome in 1955, advances have been made in understanding the key roles and functions of this organelle. The concept of lysosomal storage diseases (LSDs)—disorders characterized by aberrant, excessive storage of cellular material in lysosomes—developed following the discovery of α-glucosidase deficiency as the cause of Pompe disease in 1963. Great strides have since been made in understanding the pathobiology of LSDs and the neuronal ceroid lipofuscinoses (NCLs). The NCLs are neurodegenerative disorders that display symptoms of cognitive and motor decline, seizures, blindness, early death, and accumulation of lipofuscin in various cell types, and also show some similarities to 'classic' LSDs. Defective lysosomal storage can occur in many cell types, but the CNS and PNS are particularly vulnerable to LSDs and NCLs, being affected in two-thirds of these disorders. Most LSDs are inherited in an autosomal recessive manner, with the exception of X-linked Hunter disease, Fabry disease and Danon disease, and a variant type of adult NCL (Kuf disease). This Review provides a summary of known LSDs, and the pathways affected in these disorders. Existing therapies and barriers to development of novel and improved treatments for LSDs and NCLs are also discussed.
Key Points
-
The spectrum of lysosomal storage disease (LSD) includes defects in degradative and synthetic enzymes, lysosomal membrane defects, the neuronal ceroid lipofuscinoses (NCLs) and disorders of lysosome biogenesis and endosome–lysosome traffic
-
LSDs result in excess cellular storage and cell death in the CNS, PNS, lungs, liver, bone, skeletal and cardiac muscle and the reticuloendothelial system; symptomatology includes neurocognitive decline, seizures, blindness and hepatosplenomegaly
-
NCLs are typified by abnormal inclusions in brain, eye, liver, skin and the reticuloendothelial system, with clinical manifestations such as neurocognitive decline, blindness, seizures and early death
-
Therapies involving enzyme replacement, substrate reduction and chaperone-mediated delivery, as well as haematopoetic and other stem-cell therapies, have had modest successes in the treatment of patients with LSDs
-
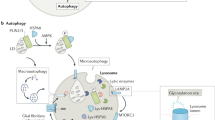
LSDs and NCLs share pathological features: abnormal lipid trafficking, dysregulation of apoptosis and autophagy, prolonged inflammation, disturbed endoplasmic reticulum–cytosol calcium balance, cellular stress, and the unfolded protein response
-
The biological underpinnings of LSDs and NCLs partly explain the limited success of 'direct' therapies for these disorders, but also provide novel targets for therapeutic approaches
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others


References
de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 7, 847–849 (2005).
Hers, H. G. Inborn lysosomal diseases. Gastroenterology 48, 625–633 (1965).
Sachs, B. A family form of idiocy, generally fatal and associated with early blindness (amaurotic family idiocy). N. Y. Med. J. 63, 697–703 (1896).
Boustany, R. M. & Mousallem, T. in Emery and Rimoin's Principles and Practice of Medical Genetics (eds Rimoin, D. et al.) 2449–2485 (Elsevier, 2007).
Ciechanover, A. Intracellular protein degradation: from a vague idea, through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim. Biophys. Acta 1824, 3–13 (2012).
Platt, F. M. & Lachmann, R. H. Treating lysosomal storage disorders: current practice and future prospects. Biochim. Biophys. Acta 1793, 737–745 (2009).
Ballabio, A. & Gieselmann, V. Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta 1793, 684–696 (2009).
Deduve, C. From cytases to lysosomes. Fed. Proc. 23, 1045–1049 (1964).
Desnick, R. J. & Schuchman, E. H. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu. Rev. Genomics Hum. Genet. 13, 307–335 (2012).
Huizing, M., Helip-Wooley, A., Westbroek, W., Gunay-Aygun, M. & Gahl, W. A. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu. Rev. Genomics Hum. Genet. 9, 359–386 (2008).
Berkovic, S. F. et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am. J. Hum. Genet. 82, 673–684 (2008).
Vogt, H. Uber familiare amaurotische idiotie und verwandte krankheitsbilder [German]. Monatsschr. Psychiatr. Neurol. 19, 161–171 (1905).
Vitner, E. B., Platt, F. M. & Futerman, A. H. Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 285, 20423–20427 (2010).
Aula, P. et al. 'Salla disease': a new lysosomal storage disorder. Arch. Neurol. 36, 88–94 (1979).
Arvio, M., Autio, S. & Louhiala, P. Early clinical symptoms and incidence of aspartylglucosaminuria in Finland. Acta Paediatr. 82, 587–589 (1993).
Frustaci, A. et al. Improvement in cardiac function in the cardiac variant of Fabry's disease with galactose-infusion therapy. N. Engl. J. Med. 345, 25–32 (2001).
Kumperscak, H. G., Paschke, E., Gradisnik, P., Vidmar, J. & Bradac, S. U. Adult metachromatic leukodystrophy: disorganized schizophrenia-like symptoms and postpartum depression in 2 sisters. J. Psychiatry Neurosci. 30, 33–36 (2005).
Arsov, T. et al. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am. J. Hum. Genet. 88, 566–573 (2011).
Stengel, O. C. Beretning om et mærkeligt Sygdomstilfaelde hos fire Sødskende I Nærheden af Röraas [Norwegian]. Eyr. Med. Tidskr. 1, 347–352 (1826).
Boustany, R. M., Alroy, J. & Kolodny, E. H. Clinical classification of neuronal ceroid-lipofuscinosis subtypes. Am. J. Med. Genet. Suppl. 5, 47–58 (1988).
Goebel, H. H., Gerhard, L., Kominami, E. & Haltia, M. Neuronal ceroid-lipofuscinosis—late-infantile or Jansky-Bielschowsky type—revisited. Brain Pathol. 6, 225–228 (1996).
Batten, F. E. Cerebral degeneration with symmetrical changes in the maculae in two members of family. Trans. Ophthalmol. Soc. UK 23, 386–390 (1903).
Vogt, H. Familiare amaurotische idiotie. Histologische and histopathologische studien [German]. Arch. Kinderheilk. 51, 1–35 (1909).
Jansky, J. Sur un cas jusqu'a présente non décrit de l'idiotie amaurotique familiale compliquée par une hypolasie du cervelet [French]. Sborn. Lek. 13, 165–196 (1908).
Bielschowsky, M. Uber spatinfantile amaurotische Idiotie mit Kleinhirnsymptomen [German]. Dtsch. Z. Nervenheilk. 50, 7–29 (1913).
Jardim, L. B., Villanueva, M. M., de Souza, C. F. & Netto, C. B. Clinical aspects of neuropathic lysosomal storage disorders. J. Inherit. Metab. Dis. 33, 315–329 (2010).
Klenk, E. Beitrage zur Chemie der Lipidosen, Niemann-Picksche und amaurotische Idiotie, Hoppe-Seyler [German]. Z. Physiol. Chem. 262, 128–143 (1939).
Svennerholm, L. The chemical structure of normal human brain and Tay-Sachs gangliosides. Biochem. Biophys. Res. Commun. 9, 436–441 (1962).
[No authors listed] Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium. Cell 82, 949–957 (1995).
Lane, S. C., Jolly, R. D., Schmechel, D. E., Alroy, J. & Boustany, R. M. Apoptosis as the mechanism of neurodegeneration in Batten's disease. J. Neurochem. 67, 677–683 (1996).
Haltia, M. & Goebel, H. H. The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim. Biophys. Acta http://dx.doi.org/10.1016/j.bbadis.2012.08.012.
Haddad, S. E. et al. CLN5 and CLN8 protein association with ceramide synthase: biochemical and proteomic approaches. Electrophoresis 33, 3798–3809 (2012).
Mole, S., Williams, R. & Goebel, H. (Eds) The Neuronal Ceroid Lipofuscinoses, Batten Disease (Oxford University Press, 2012).
Saftig, P., Beertsen, W. & Eskelinen, E. L. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy 4, 510–512 (2008).
Weaver, T. E., Na, C. L. & Stahlman, M. Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin. Cell Dev. Biol. 13, 263–270 (2002).
Saftig, P. & Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635 (2009).
Schultz, M. L., Tecedor, L., Chang, M. & Davidson, B. L. Clarifying lysosomal storage diseases. Trends Neurosci. 34, 401–410 (2011).
Braunlin, E. A. et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J. Inherit. Metab. Dis. 34, 1183–1197 (2011).
Kwon, J. Y. et al. High prevalence of carpal tunnel syndrome in children with mucopolysaccharidosis type II (Hunter syndrome). Am. J. Med. Genet. A 155A, 1329–1335 (2011).
Jurecka, A., Opoka-Winiarska, V., Jurkiewicz, E., Marucha, J. & Tylki-Szyman´ska, A. Spinal cord compression in Maroteaux-Lamy syndrome: case report and review of the literature with effects of enzyme replacement therapy. Pediatr. Neurosurg. 48, 191–198 (2012).
Pastores, G. M. et al. The MPS I registry: design, methodology, and early findings of a global disease registry for monitoring patients with mucopolysaccharidosis type I. Mol. Genet. Metab. 91, 37–47 (2007).
Guillemot, N., Troadec, C., de Villemeur, T. B., Clément, A. & Fauroux, B. Lung disease in Niemann-Pick disease. Pediatr. Pulmonol. 42, 1207–1214 (2007).
Muhlebach, M. S., Wooten, W. & Muenzer, J. Respiratory manifestations in mucopolysaccharidoses. Paediatr. Respir. Rev. 12, 133–138 (2011).
Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford) 50 (Suppl. 5), v4–v12 (2011).
Brama, I., Gay, I., Feinmesser, R. & Springer, C. Upper airway obstruction in Hunter syndrome. Int. J. Pediatr. Otorhinolaryngol. 11, 229–235 (1986).
Shih, S. L., Lee, Y. J., Lin, S. P., Sheu, C. Y. & Blickman, J. G. Airway changes in children with mucopolysaccharidoses. Acta Radiol. 43, 40–43 (2002).
Shapiro, J., Strome, M. & Crocker, A. C. Airway obstruction and sleep apnea in Hurler and Hunter syndromes. Ann. Otol. Rhinol. Laryngol. 94, 458–461 (1985).
Morehead, J. M. & Parsons, D. S. Tracheobronchomalacia in Hunter's syndrome. Int. J. Pediatr. Otorhinolaryngol. 26, 255–261 (1993).
Bonten, E. J. et al. Targeting macrophages with baculovirus-produced lysosomal enzymes: implications for enzyme replacement therapy of the glycoprotein storage disorder galactosialidosis. FASEB J. 18, 971–973 (2004).
Thappa, D. M., Singh, A., Jaisankar, T. J., Rao, R. & Ratnakar, C. Pebbling of the skin: a marker of Hunter's syndrome. Pediatr. Dermatol. 15, 370–373 (1998).
Möhrenschlager, M., Lauenstein, M., Ring, J. & Steiner, C. Synophrys. Eur. J. Med. Genet. 53, 225–226 (2010).
Spranger, J. in Emery and Rimoin's Principles and Practice of Medical Genetics (eds Rimion, D. L. et al.) 2403–2412 (Elsevier, 2007).
Eng, C. M. et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J. Inherit. Metab. Dis. 30, 184–192 (2007).
Collins, R. C., Dobkin, B. H. & Choi, D. W. Selective vulnerability of the brain: new insights into the pathophysiology of stroke. Ann. Intern. Med. 110, 992–1000 (1989).
Huebner, E. A. & Strittmatter, S. M. Axon regeneration in the peripheral and central nervous systems. Results Probl. Cell Differ. 48, 339–351 (2009).
Hoffmann, B. & Mayatepek, E. Neurological manifestations in lysosomal storage disorders—from pathology to first therapeutic possibilities. Neuropediatrics 36, 285–289 (2005).
Walkley, S. U. Pathogenic cascades in lysosomal disease—why so complex? J. Inherit. Metab. Dis. 32, 181–189 (2009).
Suvarna, J. C. & Hajela, S. A. Cherry-red spot. J. Postgrad. Med. 54, 54–57 (2008).
Birch, D. G. Retinal degeneration in retinitis pigmentosa and neuronal ceroid lipofuscinosis: an overview. Mol. Genet. Metab. 66, 356–366 (1999).
Lang, G. E. & Maumenee, I. H. in Retinal Dystrophies and Degenerations (ed. Newsome, D. A.) 319–340 (Raven Press, 1988).
Debs, R. et al. Krabbe disease in adults: phenotypic and genotypic update from a series of 11 cases and a review. J. Inherit. Metab. Dis. http://dx.doi.org/10.1007/s10545-012-9560-4.
Siddiqi, Z. A., Sanders, D. B. & Massey, J. M. Peripheral neuropathy in Krabbe disease: effect of hematopoietic stem cell transplantation. Neurology 67, 268–272 (2006).
Escolar, M. L. et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N. Engl. J. Med. 352, 2069–2081 (2005).
Kolodny, E. & Moser, H. W. in The Metabolic Basis of Inherited Disease (eds Stanbury, J. B. et al.) 881–905 (Mc-Graw-Hill, 1983).
Biegstraaten, M. et al. Small fiber neuropathy in Fabry disease. Mol. Genet. Metab. 106, 135–141 (2012).
Hannun, Y. A. & Bell, R. M. Functions of sphingolipids and sphingolipid breakdown products in cellular regulation. Science 243, 500–507 (1989).
Al Sawaf, S., Mayatepek, E. & Hoffmann, B. Neurological findings in Hunter disease: pathology and possible therapeutic effects reviewed. J. Inherit. Metab. Dis. 31, 473–480 (2008).
Tuttolomondo, A. et al. Neurological complications of Anderson-Fabry disease. Curr. Pharm. Des. [Epub ahead of print].
Neto, Â. R., Holanda, G. B., Farias, M. C., Santos da Costa, G. & Pereira, H. S. Hydrocephalus in mucopolysaccharidosis type VI successfully treated with endoscopic third ventriculostomy. J. Neurosurg. Pediatr. 11, 327–330 (2013).
Gupta, N., Rath, G. P., Bala, R., Reddy, B. K. & Chaturvedi, A. Anesthetic management in children with Hurler's syndrome undergoing emergency ventriculoperitoneal shunt surgery. Saudi J. Anaesth. 6, 178–180 (2012).
Manara, R. et al. Brain and spine MRI features of Hunter disease: frequency, natural evolution and response to therapy. J. Inherit. Metab. Dis. 34, 763–780 (2011).
Aliabadi, H. et al. Clinical outcome of cerebrospinal fluid shunting for communicating hydrocephalus in mucopolysaccharidoses I, II, and III: a retrospective analysis of 13 patients. Neurosurgery 67, 1476–1481 (2010).
Palmucci, S. et al. Imaging findings of mucopolysaccharidoses: a pictorial review. Insights Imaging http://dx.doi.org/10.1007/s13244-013-0246-8.
Jeyakumar, M., Dwek, R. A., Butters, T. D. & Platt, F. M. Storage solutions: treating lysosomal disorders of the brain. Nat. Rev. Neurosci. 6, 713–725 (2005).
Garbutt, S. & Harris, C. M. Abnormal vertical optokinetic nystagmus in infants and children. Br. J. Ophthalmol. 84, 451–455 (2000).
Sunwoo, M. K., Kim, S. M., Lee, S. & Lee, P. H. Parkinsonism associated with glucocerebrosidase mutation. J. Clin. Neurol. 7, 99–101 (2011).
Sarpong, A. et al. Protracted course of juvenile ceroid lipofuscinosis associated with a novel CLN3 mutation (p.Y199X). Clin. Genet. 76, 38–45 (2009).
Lloyd-Evans, E. et al. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 278, 23594–23599 (2003).
Tokushige, S. et al. Isolated pyramidal tract impairment in the central nervous system of adult-onset Krabbe disease with novel mutations in the GALC gene. Brain Dev. 35, 579–581 (2013).
Sehgal, R., Sharma, S., Sankhyan, N., Kumar, A. & Gulati, S. Selective corticospinal tract involvement in late-onset Krabbe disease. Neurology 77, e20 (2011).
Kamate, M. & Hattiholi, V. Predominant corticospinal tract involvement in early-onset Krabbe disease. Pediatr. Neurol. 44, 155–156 (2011).
Simonaro, C. M. et al. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc. Natl Acad. Sci. USA 107, 222–227 (2010).
Suzuki, M. et al. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann–Pick disease type C (NPC) fibroblasts: a potential basis for glial cell activation in the NPC brain. J. Neurosci. 27, 1879–1891 (2007).
Husebye, H. et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 25, 683–692 (2006).
Johnson, G. B., Brunn, G. J., Kodaira, Y. & Platt, J. L. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J. Immunol. 168, 5233–5239 (2002).
Pan, C. et al. Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood 106, 1956–1964 (2005).
Puranam, K. L., Guo, W. X., Qian, W. H., Nikbakht, K. & Boustany, R. M. CLN3 defines a novel antiapoptotic pathway operative in neurodegeneration and mediated by ceramide. Mol. Genet. Metab. 66, 294–308 (1999).
Persaud-Sawin, D. A., VanDongen, A. & Boustany, R. M. Motifs within the CLN3 protein: modulation of cell growth rates and apoptosis. Hum. Mol. Genet. 11, 2129–2142 (2002).
Persaud-Sawin, D. A. et al. Neuronal ceroid lipofuscinosis: a common pathway? Pediatr. Res. 61, 146–152 (2007).
Futerman, A. H. & van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 5, 554–565 (2004).
Domon, M. M., Besson, F., Bandorowicz-Pikula, J. & Pikula, S. Annexin A6 is recruited into lipid rafts of Niemann–Pick type C disease fibroblasts in a Ca2+-dependent manner. Biochem. Biophys. Res. Commun. 405, 192–196 (2011).
Vainio, S. et al. Defective insulin receptor activation and altered lipid rafts in Niemann–Pick type C disease hepatocytes. Biochem. J. 391, 465–472 (2005).
te Vruchte, D. et al. Accumulation of glycosphingolipids in Niemann–Pick C disease disrupts endosomal transport. J. Biol. Chem. 279, 26167–26175 (2004).
Chen, C. S., Patterson, M. C., Wheatley, C. L., O'Brien, J. F. & Pagano, R. E. Broad screening test for sphingolipid-storage diseases. Lancet 354, 901–905 (1999).
Rusyn, E., Mousallem, T., Persaud-Sawin, D. A., Miller, S. & Boustany, R. M. CLN3p impacts galactosylceramide transport, raft morphology, and lipid content. Pediatr. Res. 63, 625–631 (2008).
Persaud-Sawin, D. A. & Boustany, R. M. Cell death pathways in juvenile Batten disease. Apoptosis 10, 973–985 (2005).
Harati, H., Haliw, L., Cotman, S. & Boustany, R. M. Galactosylceramide (GalCer) as a potential treatment for juvenile neuronal ceroid lipofuscinosis (JNCL) in 13th International Conference on Neuronal Ceroid Lipofuscinosis (Batten Disease) & Patient Organisation Meeting. 54 (London, 2012).
Kobayashi, T., Shinoda, H., Goto, I., Yamanaka, T. & Suzuki, Y. Globoid cell leukodystrophy is a generalized galactosylsphingosine (psychosine) storage disease. Biochem. Biophys. Res. Commun. 144, 41–46 (1987).
Kanazawa, T. et al. Inhibition of cytokinesis by a lipid metabolite, psychosine. J. Cell Biol. 149, 943–950 (2000).
Giri, S., Khan, M., Rattan, R., Singh, I. & Singh, A. K. Krabbe disease: psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. J. Lipid Res. 47, 1478–1492 (2006).
Zaka, M. & Wenger, D. A. Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by caspase activation. Neurosci. Lett. 358, 205–209 (2004).
Nilsson, O., Månsson, J. E., Håkansson, G. & Svennerholm, L. The occurrence of psychosine and other glycolipids in spleen and liver from the three major types of Gaucher's disease. Biochim. Biophys. Acta 712, 453–463 (1982).
Kobayashi, T. et al. Accumulation of lysosphingolipids in tissues from patients with GM1 and GM2 gangliosidoses. J. Neurochem. 59, 1452–1458 (1992).
Aerts, J. M. et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl Acad. Sci. USA 105, 2812–2817 (2008).
Kolodny, E. H. & Boustany, R. M. in Hematology of Infancy and Childhood (ed. Nathan, D. O.) 1212–1247 (W. B. Saunders, 1987).
d'Azzo, A., Kolodny, E., Bonten, E. & Annunziata, I. in Nathan and Oski's Hematology of Infancy and Childhood (eds Orkin, S. et al.) 1301–1422 (W. B. Saunders, Philadelphia, 2009).
Czeh, M., Gressens, P. & Kaindl, A. M. The yin and yang of microglia. Dev. Neurosci. 33, 199–209 (2011).
DiRosario, J. et al. Innate and adaptive immune activation in the brain of MPS IIIB mouse model. J. Neurosci. Res. 87, 978–990 (2009).
Seehafer, S. S. et al. Immunosuppression alters disease severity in juvenile Batten disease mice. J. Neuroimmunol. 230, 169–172 (2011).
Pelled, D. et al. Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiol. Dis. 18, 83–88 (2005).
Korkotian, E. et al. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J. Biol. Chem. 274, 21673–21678 (1999).
Ginzburg, L., Li, S. C., Li, Y. T. & Futerman, A. H. An exposed carboxyl group on sialic acid is essential for gangliosides to inhibit calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase: relevance to gangliosidoses. J. Neurochem. 104, 140–146 (2008).
Pelled, D. et al. Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J. Biol. Chem. 278, 29496–29501 (2003).
Ginzburg, L. & Futerman, A. H. Defective calcium homeostasis in the cerebellum in a mouse model of Niemann–Pick A disease. J. Neurochem. 95, 1619–1628 (2005).
Tessitore, A. et al. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell 15, 753–766 (2004).
Kim, S. J., Zhang, Z., Hitomi, E., Lee, Y. C. & Mukherjee, A. B. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 15, 1826–1834 (2006).
Puranam, K. et al. Upregulation of Bcl-2 and elevation of ceramide in Batten disease. Neuropediatrics 28, 37–41 (1997).
Kim, S. J., Zhang, Z., Lee, Y. C. & Mukherjee, A. B. Palmitoyl-protein thioesterase-1 deficiency leads to the activation of caspase-9 and contributes to rapid neurodegeneration in INCL. Hum. Mol. Genet. 15, 1580–1586 (2006).
Dhar, S. et al. Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons. Ann. Neurol. 51, 448–466 (2002).
Zheng, Z. et al. Downregulated adaptor protein p66Shc mitigates autophagy process by low nutrient and enhances apoptotic resistance in human lung adenocarcinoma A549 cells. FEBS J. http://dx.doi.org/10.1111/febs.12416.
B'chir, W. et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. http://dx.doi.org/10.1093/nar/gkt563.
Su, M., Mei, Y. & Sinha, S. Role of the crosstalk between autophagy and apoptosis in cancer. J. Oncol. 2013, 102735 (2013).
Müller, S., Dennemärker, J. & Reinheckel, T. Specific functions of lysosomal proteases in endocytic and autophagic pathways. Biochim. Biophys. Acta 1824, 34–43 (2012).
Maiuri, M. C., Zalckvar, E., Kimchi, A. & Kroemer, G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752 (2007).
Raben, N., Roberts, A. & Plotz, P. H. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 26, 45–48 (2007).
Alvarez, V. E., Niemirowicz, G. T. & Cazzulo, J. J. The peptidases of Trypanosoma cruzi: digestive enzymes, virulence factors, and mediators of autophagy and programmed cell death. Biochim. Biophys. Acta 1824, 195–206 (2012).
Pacheco, C. D. & Lieberman, A. P. Lipid trafficking defects increase Beclin-1 and activate autophagy in Niemann–Pick type C disease. Autophagy 3, 487–489 (2007).
Chang, J. W., Choi, H., Cotman, S. L. & Jung, Y. K. Lithium rescues the impaired autophagy process in CbCln3Δex7/8/Δex7/8 cerebellar cells and reduces neuronal vulnerability to cell death via IMPase inhibition. J. Neurochem. 116, 659–668 (2011).
Settembre, C., Fraldi, A., Rubinsztein, D. C. & Ballabio, A. Lysosomal storage diseases as disorders of autophagy. Autophagy 4, 113–114 (2008).
US National Library of Medicine. ClinicalTrials.gov [online], (2013).
US National Library of Medicine. ClinicalTrials.gov [online], (2013).
US National Library of Medicine. ClinicalTrials.gov [online], (2013).
US National Library of Medicine. ClinicalTrials.gov [online], (2012).
US National Library of Medicine. ClinicalTrials.gov [online], (2012).
US National Library of Medicine. ClinicalTrials.gov [online], (2013).
US National Library of Medicine. ClinicalTrials.gov [online], (2012).
US National Library of Medicine. ClinicalTrials.gov [online], (2012).
US National Library of Medicine. ClinicalTrials.gov [online], (2013).
Hirabayashi, Y., Merrill, A. H. Jr & Shayman, J. A. in Sphingolipid Biology (eds Hirabayashi, Y. et al.) 83–94 (Springer, 2006).
Valayannopoulos, V. & Wijburg, F. A. Therapy for the mucopolysaccharidoses. Rheumatology (Oxford) 50 (Suppl. 5), v49–v59 (2011).
Urbanelli, L. et al. Therapeutic approaches for lysosomal storage diseases: a patent update. Recent Pat. CNS Drug Discov. 8, 91–109 (2013).
Platt, F. M. & Jeyakumar, M. Substrate reduction therapy. Acta Paediatr. Suppl. 97, 88–93 (2008).
Maegawa, G. H. et al. Substrate reduction therapy in juvenile GM2 gangliosidosis. Mol. Genet. Metab. 98, 215–224 (2009).
Shapiro, B. E., Pastores, G. M., Gianutsos, J., Luzy, C. & Kolodny, E. H. Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet. Med. 11, 425–433 (2009).
Young-Gqamana, B. et al. Migalastat HCl reduces globotriaosylsphingosine (lyso-Gb3) in Fabry transgenic mice and in the plasma of Fabry patients. PLoS ONE 8, e57631 (2013).
US National Library of Medicine. ClinicalTrials.gov [online], (2012).
Lukina, E. et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: 2-year results of a phase 2 study. Blood 116, 4095–4098 (2010).
Rountree, J. S. et al. Design, synthesis, and biological evaluation of enantiomeric β-N-acetylhexosaminidase inhibitors LABNAc and DABNAc as potential agents against Tay-Sachs and Sandhoff disease. ChemMedChem 4, 378–392 (2009).
Tropak, M. B. et al. A sensitive fluorescence-based assay for monitoring GM2 ganglioside hydrolysis in live patient cells and their lysates. Glycobiology 20, 356–365 (2010).
Escolar, M. L., Poe, M. D., Martin, H. R. & Kurtzberg, J. A staging system for infantile Krabbe disease to predict outcome after unrelated umbilical cord blood transplantation. Pediatrics 118, e879–e889 (2006).
de Ru, M. H. et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J. Rare Dis. 6, 55 (2011).
Kohan, R. et al. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr. Pharm. Biotechnol. 12, 867–883 (2011).
Worgall, S. et al. Neurological deterioration in late infantile neuronal ceroid lipofuscinosis. Neurology 69, 521–535 (2007).
Crystal, R. G. et al. Clinical protocol. Administration of a replication-deficient adeno-associated virus gene transfer vector expressing the human CLN2 cDNA to the brain of children with late infantile neuronal ceroid lipofuscinosis. Hum. Gene Ther. 15, 1131–1154 (2004).
Sands, M. S. & Davidson, B. L. Gene therapy for lysosomal storage diseases. Mol. Ther. 13, 839–849 (2006).
US National Library of Medicine. ClinicalTrials.gov [online], (2013).
US National Library of Medicine. ClinicalTrials.gov [online], (2012).
US National Library of Medicine. ClinicalTrials.gov [online], (2013).
Brooks, D. A., Muller, V. J. & Hopwood, J. J. Stop-codon read-through for patients affected by a lysosomal storage disorder. Trends Mol. Med. 12, 367–373 (2006).
Zhang, Z. et al. Lysosomal ceroid depletion by drugs: therapeutic implications for a hereditary neurodegenerative disease of childhood. Nat. Med. 7, 478–484 (2001).
Ma, X. et al. Improvements in mucopolysaccharidosis I mice after adult retroviral vector-mediated gene therapy with immunomodulation. Mol. Ther. 15, 889–902 (2007).
Jakóbkiewicz-Banecka, J., Piotrowska, E., Narajczyk, M., Baran´ska, S. & Wegrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis, which corrects storage in cells of patients suffering from mucopolysaccharidoses, acts by influencing an epidermal growth factor-dependent pathway. J. Biomed. Sci. 16, 26 (2009).
Xu, M. et al. δ-Tocopherol reduces lipid accumulation in Niemann–Pick type C1 and Wolman cholesterol storage disorders. J. Biol. Chem. 287, 39349–39360 (2012).
Choudhury, A. et al. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann–Pick C cells. J. Clin. Invest. 109, 1541–1550 (2002).
Davidson, C. D. et al. Chronic cyclodextrin treatment of murine Niemann–Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 4, e6951 (2009).
Kirkegaard, T. et al. Hsp70 stabilizes lysosomes and reverts Niemann–Pick disease-associated lysosomal pathology. Nature 463, 549–553 (2010).
Zhou, Q. H., Boado, R. J., Lu, J. Z., Hui, E. K. & Pardridge, W. M. Brain-penetrating IgG-iduronate 2-sulfatase fusion protein for the mouse. Drug Metab. Dispos. 40, 329–335 (2012).
Maga, J. A. et al. Glycosylation-independent lysosomal targeting of acid alpha-glucosidase enhances muscle glycogen clearance in pompe mice. J. Biol. Chem. 288, 1428–1438 (2013).
Montano, A. M. et al. Acidic amino acid tag enhances response to enzyme replacement in mucopolysaccharidosis type VII mice. Mol. Genet. Metab. 94, 178–189 (2008).
Koike, M. et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am. J. Pathol. 167, 1713–1728 (2005).
Repnik, U., Stoka, V., Turk, V. & Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 1824, 22–33 (2012).
Staretz-Chacham, O., Lang, T. C., LaMarca, M. E., Krasnewich, D. & Sidransky, E. Lysosomal storage disorders in the newborn. Pediatrics 123, 1191–1207 (2009).
Smith, K. R. et al. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 22, 1417–1423 (2013).
US National Library of Medicine. ClinicalTrials.gov [online], (2013).
Ghosh, A., Corbett, G. T., Gonzalez, F. J. & Pahan, K. Gemfibrozil and fenofibrate, Food and Drug Administration-approved lipid-lowering drugs, up-regulate tripeptidyl-peptidase 1 in brain cells via peroxisome proliferator-activated receptor α: implications for late infantile Batten disease therapy. J. Biol. Chem. 287, 38922–38935 (2012).
Miller, J. N., Chan, C. H. & Pearce, D. A. The role of nonsense-mediated decay in neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 22, 2723–2734 (2013).
Eliyahu, E. et al. Anti-TNF-alpha therapy enhances the effects of enzyme replacement therapy in rats with mucopolysaccharidosis type VI. PLoS ONE 6, e22447 (2011).
Coutinho, M. F., Prata, M. J. & Alves, S. Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol. Genet. Metab. 105, 542–550 (2012).
Koeberl, D. D. et al. Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol. Genet. Metab. 103, 107–112 (2011).
Li, S. et al. Adjunctive β2-agonists reverse neuromuscular involvement in murine Pompe disease. FASEB J. 27, 34–44 (2013).
Messinger, Y. H. et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet. Med. 14, 135–142 (2012).
Abbott, M. A. et al. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol. Genet. Metab. 104, 583–586 (2011).
Jmoudiak, M. & Futerman, A. H. Gaucher disease: pathological mechanisms and modern management. Br. J. Haematol. 129, 178–188 (2005).
Lake, B. D., Young, E. P. & Winchester, B. G. Prenatal diagnosis of lysosomal storage diseases. Brain Pathol. 8, 133–149 (1998).
Summary of Gaucher Disease Patient Prioritization For Treatment Recommendations From The Meeting Of Gaucher Treatment Experts, September 9th, 2009, Chicago, IL, USA. Gaucher Disease Organization [online] (2009).
Brady, R. O. Enzyme replacement for lysosomal diseases. Annu. Rev. Med. 57, 283–296 (2006).
Bailey, L. An overview of enzyme replacement therapy for lysosomal storage diseases. Online J. Issues Nurs. 13, manuscript 3 (2008).
Guo, W. X., Mao, C., Obeid, L. M. & Boustany, R. M. A disrupted homologue of the human CLN3 or juvenile neuronal ceroid lipofuscinosis gene in Saccharomyces cerevisiae: a model to study Batten disease. Cell. Mol. Neurobiol. 19, 671–680 (1999).
Chattopadhyay, S., Muzaffar, N. E., Sherman, F. & Pearce, D. A. The yeast model for batten disease: mutations in btn1, btn2, and hsp30 alter pH homeostasis. J. Bacteriol. 182, 6418–6423 (2000).
Boustany, R. M., Shareef, I. & El-Haddad, S. in Emery and Rimoin's Principles and Practice of Medical Genetics Ch. 109 (Elsevier, 2013).
Auffray, C. et al. Genome Medicine: past, present and future. Genome Med. 3, 6 (2011).
Khoury, M. J. Public health genomics: 15 years on. Centers for Disease Control and Prevention [online], (2013).
Benitez, B. A. et al. Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS ONE 6, e26741 (2011).
Smith, K. R. et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage Am. J. Hum. Genet. 90, 1102–1107 (2012).
Staropoli, J. F. et al. A homozygous mutation in KCTD7 links neuronal ceroid lipofuscinosis to the ubiquitin-proteasome system. Am. J. Hum. Genet. 91, 202–208 (2012).
Acknowledgements
The author is grateful to H. Maacaron for her valuable assistance with the referencing of this manuscript.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
R. N. Boustany is an inventor on the following US patent applications: 8,003,327 (methods of screening for risk of proliferative disease and methods for the treatment of proliferative disease); 6,821,995 (a method of treating Batten disease); and 8,242,086 (methods and compositions for treating disorders caused by deficiency in a gene product of a CLN gene).
Rights and permissions
About this article
Cite this article
Boustany, RM. Lysosomal storage diseases—the horizon expands. Nat Rev Neurol 9, 583–598 (2013). https://doi.org/10.1038/nrneurol.2013.163
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneurol.2013.163
This article is cited by
-
Innate immune sensing of lysosomal dysfunction drives multiple lysosomal storage disorders
Nature Cell Biology (2024)
-
A computational approach to analyzing the functional and structural impacts of Tripeptidyl-Peptidase 1 missense mutations in neuronal ceroid lipofuscinosis
Metabolic Brain Disease (2024)
-
CLN3 is required for the clearance of glycerophosphodiesters from lysosomes
Nature (2022)
-
Fabry Disease with Aseptic Meningitis: A Case Series and Literature Review of an Underestimated Clinical Presentation
Current Medical Science (2022)
-
Zinc supplementation improves the harvest purity of β-glucuronidase from CHO cell culture by suppressing apoptosis
Applied Microbiology and Biotechnology (2020)
