
Abstract
Mild cognitive impairment (PD-MCI) and dementia (PDD) are among the most frequent non-motor symptoms in Parkinson’s disease (PD). PD-MCI is six times more likely than age-matched controls to develop dementia and the PDD prevalence is 80% after 15–20 years of disease. Therefore, research has focused on the identification of early dementia biomarkers including specific cognitive at-risk profiles hoping to implement therapeutic interventions when they are most likely to be efficacious. However, given the heterogeneous neuropathological, neurochemical, and neuropsychological nature of cognitive deficits, definition of a comprehensive cognitive model of PDD is a challenge. Evidence from neuroimaging studies using different methods and techniques suggests that in addition to degeneration of the dopaminergic system, other mechanisms have a role including β-amyloid and tau deposition, and that specific cognitive scales could help identifying a malignant profile. Prospective studies combining neuroimaging techniques and specific cognitive tests are required to define the interplay between the various neurodegenerative processes and the contribution of structural disconnection in brain functional networks, heralding the development of dementia in PD.
Similar content being viewed by others


Introduction
Diagnosis of Parkinson’s disease (PD) relies on the presence of specific motor symptoms such as bradykinesia, resting tremor, rigidity, asymmetric manifestations, and good response to levodopa.1 PD patients also present several non-motor features often preceding motor signs by many years with a negative impact on quality of life, increased caregiver burden, and annual economic costs (estimated at ~14 billion in Europe).2 In particular, management of cognitive impairment is an area of growing clinical interest given its heterogeneous manifestations and the risk of dementia.3 Considering that up to 80% of PD patients after 15–20 years of disease have progressed to dementia,4 there is urgent need to identify early biomarkers to establish efficacious treatments and monitor cognitive deterioration. However, the presence of heterogeneous underlying pathophysiology and neuropsychological phenotype makes it difficult to define a unique model explaining the whole cognitive picture. Indeed, PD neuropathology demonstrates Lewy body-type degeneration in cortical and limbic structures,5 coexisting Alzheimer pathology (neurofibrillary tangle and senile plaques)6 as well as microvascular lesions with the involvement of subcortical structures.7 A post-mortem study showed that only 38% of PD patients with dementia (PDD) had “pure” or exclusive Lewy body pathology, whereas 59% presented combined Lewy body and β-amyloid plaques, and 3% concomitant Lewy body, β-amyloid plaques, and neurofibrillary tangles.8,9 Moreover, there is a significant positive relationship between cortical β-amyloid deposition and cognitive impairment,10 and β-amyloid accumulation is detected in one-third of non-demented PD.11 By contrast, only few neuropathology studies are available in Parkinson with mild cognitive impairment (PD-MCI) but results are heterogeneous.12,13
In addition, the coexistence of overlapping pathologies does not always match with the clinical manifestations due to the various cerebral distribution of abnormal protein deposits.14 This complex framework occurs in the context of multiple neurotransmitter deficiencies (including dopamine, noradrenaline, serotonin, and acetylcholine) and presence of genes that increase the dementia risk (such as ɑ-synuclein mutation (SNCA), the apolipoprotein ε4 (APOE4) allele, and the microtubule-associated protein tau (MAPT) H1 haplotype7). Finally, an approach based only on the clinical symptoms would be insufficient to define PD cognitive environment, given the poor sensitivity of the MCI diagnosis guidelines published in literature.15
This review outlines the most important issues related to the nature of the neuropsychological deficits, the identification of PD cognitive profile by a clinical semi-structured interview, and highlights the potential role of neuroimaging techniques such as magnetic resonance imaging, positron emission tomography, and single-photon emission computed tomography to increase the accuracy of early detection of cognitive dysfunction.
Cognitive deficits in PD: the clinical neuropsychology-based approach
After an initial controversy about the presence of cognitive deficits,16 PD cognitive impairment and dementia are now well documented, and listed among the most prevalent and disabling non-motor symptoms.
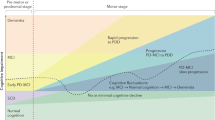
Initially (1980–1990), cognitive deficits were considered executive in nature due to the dopamine depletion in the nigro-striatal pathways. This hypothesis was confirmed by several studies showing impairment in working memory, planning/sequencing (as in then Tower of London test), task-switching (as in the Wisconsin Card Sort test), response inhibition process (as in the Stroop task), memory recall, verbal fluency as well as many aspect of motor cognition (as psychomotor speed).17 They can be detected in early untreated patients, or even at premotor stage and worsen with disease progression.18 Moreover, evidence suggests spatiotemporal asymmetry of dopamine depletion at the striatal level characterized by an early involvement of the dorsolateral prefrontal circuit progressively extending to the orbito-frontal pathway involved in reward-based learning.19 This explains the non-linear effect on cognition of dopaminergic drugs. In line with the ‘dopamine overdose hypothesis’, dopaminergic medications may ameliorate dorsolateral prefrontal cortex-related executive functions, but overdose the intact portion with detrimental effect on orbito-frontal cortex-related executive functions such as decision-making and reversal learning tasks. In addition, as levodopa treatment only partially restores cognitive deficits,20 it is evident that a proportion of the impairment was not dopamine-related and neurotransmitter systems such as acetylcholine, noradrenaline, and serotonin are involved in cognition. More specifically, this regards specific attention problems (such as impaired vigilance with fluctuating level of alertness),21 memory deficits (with free recall retrieval more prominent at the earlier stage of the disease22 and an additional storage component memory present in PDD), and subtle visuo-spatial and perceptive impairments (difficulties with the perception of extra personal space23 and in recognizing objects based on their form).24 Unsolved questions regard the nature, dissociation, and progression of language and semantic impairments particularly syntactic, action-verb, and action semantic skills. Some authors posit that these deficits are epiphenomenal of executive dysfunction or impairment of selective attention or even working memory deficits,25 whereas others showed the presence of specific linguistic deficits (action-naming) without concurrent executive defects.26 Interestingly, a recent study found linguistic deficits in PD without MCI. In particular, action-naming and action-association were disrupted in the absence of executive deficits, suggesting a sui generis defect present since early disease stages.27
On the basis of the heterogeneity of cognitive deficits and cross-sectional and longitudinal evidence showing that visuo-spatial and semantic memory impairments are highly sensitive in detecting the transition to PDD,28–30 the ‘dual syndrome hypothesis’ was proposed. This hypothesis is based on the existence of two independent, partially overlapping syndromes in PD: (a) a frontal-striatal network dysfunction present at the early stage of the disease, which is dopamine modulated, leading to deficit in working memory, attention, planning, and response inhibition; (b) an additional more posterior cortical degeneration, which is associated with cholinergic loss and wherever present would lead to dementia.31 A major question regards the relationship between these two syndromes and more specifically on how many PD-MCI cases proceed to clinical dementia and what degree of overlap exists between these early deficits and the prevalence of dementia.
How to cluster cognitive deficits
The concept of MCI has been applied in PD to improve early dementia detection and is used as an umbrella term to describe the heterogeneous deficits observed in the range between normal cognition and PDD.15 Prospective longitudinal studies showed that mild cognitive deficits can be detected in ~15–25% of newly diagnosed patients28,32 and could be present even before motor symptoms appear.17,33 PD-MCI patients have an annual rate of dementia between 9 and 15%,34 and clinical and demographic variables (mainly age, disease duration, and disease severity) affect the incidence.35 However, the low specificity of the MCI concept has contributed to this variability mainly because there are methodological differences in classification criteria (such as abnormal threshold range from −1 to −2 s.d.’s), there is no consensus about which and how many cognitive tests are needed for cognitive profiling and the cut-off scores are derived from inadequate normative data.36–39 For example, the threshold values indicated by the MCI Movement Disorder Task Force guideline do not place patient’s cognitive impairment in the continuum between PD without cognitive deficits and dementia, but can only be applied as dichotomous criteria for exclusion/inclusion in a given cognitive state. Varying those thresholds for the definition of PD-MCI affects frequency and clinical profile of MCI.40 Specific and more sensitive cut-off scores should be adopted for each test to increase accuracy of cognitive diagnosis. Our group previously observed that screening and diagnostic cut-off values for tests identified as good discriminator of PD-MCI would lie within the healthy subject range according to the published normative data, underling the risk of false-negative error in making MCI diagnosis.41 When Petersen42 and Winblad43 MCI subtype classification is applied to PD, it emerges that the non-amnestic single domain impairment predominates in PD-MCI44 and only few patients exhibit a more posterior cortical profile.45 Moreover, it is still uncertain which cognitive pattern best predicts the progression to PDD,46,47 although evidence suggests that impairment in semantic fluency and intersecting pentagon copying may be very sensitive.35 We recently assessed the discrimination power of several neuropsychological tests in differentiating cognitive stages in 105 PD patients (Parkinson without cognitive deficits versus PD-MCI versus PDD). We observed that attention/set-shifting, semantic memory, and language (category fluency task, naming test, prose memories, and similarities) as well as visuo-spatial functions (clock drawing test) were best in discriminating PD-MCI patients and an additional involvement of posterior cognitive functions (visuo-spatial, visuo-perceptive abilities together with language) in PDD,28 similar to that observed in Alzheimer disease.48
Overall, these findings, although heterogeneous, are in line with the presence of two cognitive profiles in PD-MCI associated with different aetiologies: (1) frontal-striatal-based executive–attention deficits in which the dopamine depletion has an important role; (2) posterior alterations49 associated with the increased risk of progression to PDD, presence of β-amyloid abnormalities, and further depletion of non-dopaminergic neurotransmitters. Finally, there is also evidence that PD cognition may show a non-linear decline depending on the impaired function.50,51
So far, longitudinal studies using PD-MCI published criteria demonstrated that their application may not be ideal in identifying the high dementia-risk PD cognitive profile likely because they do not take into account the contribution of single test deficits in predicting dementia.
The neuroimaging evidence
Because of the aforementioned issues associated with the MCI concept in PD, almost 20 years neuroimaging studies based on PD-MCI/PDD published criteria, did not clarify the issue. Moreover, inclusion of small and poorly characterized subjects, heterogeneous methodological approaches (imaging preprocessing steps, motion artefacts, and limitation of specific techniques (voxel-based morphometry versus cortical thickness) that may be not high sensitive to detect cognitive decline at early PD stage) and medication status (on/off state) have increased variability in study findings.
Neuroimaging approaches have attempted to identify that brain components and correlated cognitive performance characterize the prodromal dementia profile (see Table 1 for a summary of the main studies) and discuss the contribution of anatomical ([gray (GM) and white matter (WM)]) abnormalities as markers of cognitive dysfunctions showing evidence of both frontal and posterior functional connectivity changes as possible biomarkers.
Structural brain alterations
The PD-MCI structural phenotype emerging from studies using regions of interest analysis, voxel-based morphometry, and cortical thickness is heterogeneous.52,53,55 There is consensus on the presence of widespread cortical atrophy as well as increased and decreased thickness in neocortex and subcortical regions in PDD.49,56 Interestingly, impairment in specific domains correlates with both anterior and posterior cortical thinning in PD-MCI,57 underling the notion of widespread and shared brain network recruitment for high-level cognitive functions. Two longitudinal studies found a pattern of accelerated cortical thinning in PD non-demented compared with controls, in spite of similar controls cortical thickness pattern at baseline.58,59 However, these studies had limitations due to sample size59 and heterogeneous cohort.58 Conversely, a recent 2-year longitudinal study showed lower GM density at baseline in the prefrontal and insular cortex, and caudate nuclei in PD-MCI subjects who developed dementia.54 In the same line, 18 months’ longitudinal study showed bilateral temporal thinning at baseline in PD converters.60
Overall, these studies are in line with the dual syndrome hypothesis and support the concept of progressive temporo-parietal thinning together with frontal atrophy as a biomarker for progression towards dementia. It is still to be understood how the current PD-MCI categorization should be revised to achieve a finer delineation of the subgroups.
White Matter alterations
Findings of WM microstructures across the full spectrum of cognitive status in PD are more consistent and a unique pattern of diffusivity changes underling impairment in distinct cognitive domains have been reported.61,62 Carlesimo et al.66 showed diffusion abnormalities correlating with memory deficits, in the posterior cingulated and hippocampus in non-demented PD, even in the absence of volume loss. Agosta et al.64 found structural WM alterations mainly located in the frontal regions in PD-MCI versus healthy controls, in spite of no GM atrophy. A recent study conducted in early PD showed increased regional mean diffusivity (MD) compared with healthy controls in frontal and parietal tracts correlating with semantic fluency and tower of London task, in the absence of reduction in both fractional anisotropy (FA) and GM volume.65 Indeed, increased MD, correlating with cognitive deficits, has been seen together with minimal FA alterations and absence of GM atrophy in both AD or PD.63,76 Taken together, these findings show that increased MD may be an early-stage marker of cognitive decline, and it could be detected before reductions of either FA and GM volume.
Given these consistent WM findings, some authors posit that PD could be primarily the result of synaptic dysfunctions (due to the presence of Lewy neurites in the axon), which subsequently lead to cell death.11,77 This evidence underlies the potential role of DTI as preclinical dementia biomarker in the absence of atrophic changes.
Resting-state and functional connectivity studies
In the context of an integrated model of brain function, neuroimaging studies in healthy subjects showed that coherent and reliable pattern of spontaneous neuronal oscillation are observed at rest.78 Analyzing the correlated and anti-correlated regions activity, it has been observed that cognitive processing is subserved by a large-scale intrinsic connectivity networks (ICNs).79 In particular, the default mode network (DMN),80 the dorsal–ventral attention network (DAN),81 the fronto-parietal network (FPN),82 the central–executive network (CEN),83 the visuo-spatial, and the salience network (SN) has been extensively investigated.67 The study of these ICNs suggested that task-free or resting-state techniques are a useful tool to better understand human behavior and diseases.84
Previous studies suggested that resting-state brain organization could provide insight on how the brain reacts during external tasks.85 For example, dysfunctional connectivity among the SN, CEN, and DMN has been in several brain disorders,86 including PD.73,87 In particular, Putcha et al.87 found an aberrant positive coupling between DMN and right CEN, compared with the expected anti-correlation seen in HC,88 possibly reflecting a failure to suppress DMN activity or to modulate top-down signals between DMN and CEN.89 They also found a reduction in functional coupling between SN and right CEN in PD when compared with HC. Insula and dorsal anterior cingulated cortex, key nodes of the SN,83 are anatomically and functionally connected with CEN.90 Interestingly, network changes may reflect an increased PD-related pathological burden in the striatum and insula as suggested by Christopher et al.,74 who found that combined striatal-insula dopamine depletion predicted executive dysfunction in PD-MCI. The role of the fronto-insular cortex has been recently recognized by the finding of reduced functional connectivity in right fronto-insular regions and the DAN (which was correlated with attention and executive network) in treated PD-MCI.68 Combined with evidence that dopamine modulates resting-state patterns of coupling between DMN–FPN–DAN networks,91 it can be inferred that ICNs changes mediated by insular dopaminergic denervation have a role in dopamine modulated fronto-striatal deficits.92 These results, corroborate the presence of a frontal syndrome underlying the association between ICNs changes and dopamine-related cognitive deficits in PD.69 However, other studies observed the presence of posterior functional connectivity changes in PD.72 Tessitore et al.70 reported decreased functional connectivity of the right medial temporal lobe which, projecting to the hippocampus, predicted memory performance, and decreased inferior parietal cortex, which predicted visual-spatial scores, in cognitively unimpaired PD patients on medication. Baggio et al.68 observed increased connectivity between DMN and posterior cortical regions that were also atrophic. Moreover, ICN changes were associated with visual-perceptual deficits. A longitudinal study,71 using a synchronization likelihood as a measure of coupling, observed resting-state functional connectivity reductions mainly involving posterior cortical regions in PD-MCI on therapy. Interestingly, those functional connectivity changes correlated with cognitive decline. Other studies reported greater cholinergic denervation in PD with dementia compared with PD75 and the synergic role of acetylcholine and dopamine activity in predicting cognitive performance.93 In summary, current evidence on functional connectivity highlights that both frontal and posterior syndromes have resting-state correlates, with posterior alterations detectable also at preclinical cognitive stage and most likely predictor of worse cognitive outcome. Indeed, fronto-striatal executive deficits are mainly mediated by dopaminergic alterations, whereas additional neurotransmitters depletion, including cholinergic deficits, are associated with widespread cortical atrophy.31,45
Discussion and Conclusions
Dementia in PD causes significant disability and leads to institutionalization and death. Early identification of patients at risk for early dementia is required to properly apply advanced treatments such as deep brain stimulation and design, and implement therapeutic interventions early when they are most likely to have efficacy. In the present review, we have shown the complexity in providing a comprehensive cognitive model of PDD, given the neuropathological, neurochemical, and neuropsychological heterogeneity of cognitive profiles. Loss of nigro-striatal dopamine neurons, cholinergic projections, limbic, and cortical alpha-synuclein positive Lewy bodies are associated with motor and cognitive deterioration.5,11 Although the presence of two coexisting cognitive profiles in PD is consistently reported across studies, the aforementioned evidence also suggests that each clinical, neuropsychological, and magnetic resonance imaging assessments cannot by themselves reliably discriminate PD patients who are likely to progress to dementia. Moreover, the presence of posterior functional connectivity abnormalities in cognitively unimpaired patients70 casts doubts about the temporal aspect of the dual syndrome hypothesis, which would postulate initial frontal–executive abnormalities followed by a subsequent additional posterior deficits. The confounding factors could be the a priori cognitive categorizations in PD-MCI and PDD, adopting heterogeneous approaches to classify patients without taking into account the temporal onset of cognitive features in respect of motor symptoms. Indeed, defining the timing of cognitive decline in PD is important and needs to be clarified. Intriguingly, neuropathology studies give us an idea of the importance of defining the onset of cognitive deficits. PD, PDD, and dementia with Lewy bodies (DLB) patients share the presence of α-synuclein aggregates in Lewy bodies and neuritis, and different timing in the onset of cognitive and motor manifestations may reflect the diverse regional burden and cerebral distribution of the pathology. Moreover, β-amyloid deposition is a frequent feature of DLB strongly affecting clinical manifestations.94 In PDD, duration of parkinsonism before dementia is associated with different patterns of brain pathology and neurochemical abnormalities.95 Similarly, retrospective data from the UK Brain Bank demonstrated that very long disease duration (up to 28 years) showed no clinical dementia at death irrespective of the presence of cortical Lewy bodies similar to that observed in DLB.9
In conclusion, a single model for Lewy body cognitive abnormalities and dementia should be adopted. In particular, prospective studies combining neuroimaging and sensitive cognitive tests should clarify the interplay between the various neurodegenerative processes, and the contribution of structural disconnections in brain functional networks, heralding the development of dementia. We suggest that additional pathology has a role in the early development of dementia and this includes β-amyloid deposition possibly in specific brain regions. Finally, the mini-mental state examination, a general cognitive scale widely used in AD, which is relatively insensitive in detecting cognitive abnormalities in the early phases of PD, but it shows a rapid decline (yearly point loss of 7–8%) in the presence of dementia,28,96 may be a valuable cognitive biomarker to identify patients at risk for dementia.
References
Berardelli, A. et al. EFNS/MDS-ES/ENS [corrected] recommendations for the diagnosis of Parkinson's disease 1. Eur. J. Neurol. 20, 16–34 (2013).
Olesen, J., Gustavsson, A., Svensson, M., Wittchen, H. U. & Jonsson, B. The economic cost of brain disorders in Europe 8. Eur. J. Neurol. 19, 155–162 (2012).
Pagonabarraga, J. & Kulisevsky, J. Cognitive impairment and dementia in Parkinson's disease. Neurobiol. Dis. 46, 590–596 (2012).
Aarsland, D. & Kurz, M. W. The epidemiology of dementia associated with Parkinson's disease. Brain Pathol. 20, 633–639 (2010).
Braak, H., Rub, U., Jansen Steur, E. N., Del, T. K. & de Vos, R. A. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64, 1404–1410 (2005).
Irwin, D. J. et al. Neuropathologic substrates of Parkinson disease dementia. Ann. Neurol. 72, 587–598 (2012).
Halliday, G. M., Leverenz, J. B., Schneider, J. S. & Adler, C. H. The neurobiological basis of cognitive impairment in Parkinson's disease. Mov. Disord. 29, 634–650 (2014).
Kotzbauer, P. T. et al. Pathologic accumulation of alpha-synuclein and Abeta in Parkinson disease patients with dementia. Arch. Neurol. 69, 1326–1331 (2012).
Colosimo, C., Hughes, A. J., Kilford, L. & Lees, A. J. Lewy body cortical involvement may not always predict dementia in Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 74, 852–856 (2003).
Petrou, M. et al. Abeta-amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology 79, 1161–1167 (2012).
Petrou, M. et al. Amyloid deposition in Parkinson's disease and cognitive impairment: a systematic review. Mov. Disord. 30, 928–935 (2015).
Adler, C. H. et al. Heterogeneous neuropathological findings in Parkinson's disease with mild cognitive impairment. Acta Neuropathol. 120, 827–828 (2010).
Jellinger, K. A. Mild cognitive impairment in Parkinson disease: heterogenous mechanisms. J. Neural Transm. (Vienna) 120, 157–167 (2013).
Galvin, J. E., Pollack, J. & Morris, J. C. Clinical phenotype of Parkinson disease dementia. Neurology 67, 1605–1611 (2006).
Litvan, I. et al. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force guidelines. Mov. Disord. 27, 349–356 (2012).
Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 14, 223–236 (2002).
Goldman, J. G., Williams-Gray, C., Barker, R. A., Duda, J. E. & Galvin, J. E. The spectrum of cognitive impairment in Lewy body diseases. Mov. Disord. 29, 608–621 (2014).
Santangelo, G. et al. Mild cognitive impairment in newly diagnosed Parkinson's disease: a longitudinal prospective study. Parkinsonism Relat. Disord. 21, 1219–1226 (2015).
Cools, R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson's disease. Neurosci. Biobehav. Rev. 30, 1–23 (2006).
Lange, K. W. et al. L-DOPA withdrawal in Parkinson's disease selectively impairs cognitive performance in tests sensitive to frontal lobe dysfunction. Psychopharmacology (Berl) 107, 394–404 (1992).
Ballard, C. G. et al. Fluctuations in attention: PD dementia vs DLB with parkinsonism. Neurology 59, 1714–1720 (2002).
Costa, A. et al. Free and cued recall memory in Parkinson's disease associated with amnestic mild cognitive impairment. PLoS.One 9, e86233 (2014).
Montse, A., Pere, V., Carme, J., Francesc, V. & Eduardo, T. Visuospatial deficits in Parkinson's disease assessed by judgment of line orientation test: error analyses and practice effects. J. Clin. Exp. Neuropsychol. 23, 592–598 (2001).
Kida, Y., Tachibana, H., Takeda, M., Yoshikawa, H. & Okita, T. Recognition memory for unfamiliar faces in Parkinson's disease: behavioral and electrophysiologic measures. Parkinsonism Relat. Disord. 13, 157–164 (2007).
Bastiaanse, R. & Leenders, K. L. Language and Parkinson's disease. Cortex 45, 912–914 (2009).
Ibanez, A. et al. Motor-language coupling: direct evidence from early Parkinson's disease and intracranial cortical recordings. Cortex 49, 968–984 (2013).
Bocanegra, Y. et al. Syntax, action verbs, action semantics, and object semantics in Parkinson's disease: Dissociability, progression, and executive influences. Cortex 69, 237–254 (2015).
Biundo, R. et al. Cognitive profiling of Parkinson disease patients with mild cognitive impairment and dementia. Parkinsonism Relat. Disord. 20, 394–399 (2014).
Williams-Gray, C. H. et al. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson's disease. J. Neurol. 256, 493–498 (2009).
Hobson, P. & Meara, J. Mild cognitive impairment in Parkinson's disease and its progression onto dementia: a 16-year outcome evaluation of the Denbighshire cohort. Int. J. Geriatr. Psychiatry 30, 1048–1055 (2015).
Kehagia, A. A., Barker, R. A. & Robbins, T. W. Cognitive impairment in Parkinson's disease: the dual syndrome hypothesis. Neurodegener. Dis. 11, 79–92 (2013).
Aarsland, D., Bronnick, K., Larsen, J. P., Tysnes, O. B. & Alves, G. Cognitive impairment in incident, untreated Parkinson disease: the Norwegian ParkWest study. Neurology 72, 1121–1126 (2009).
Pigott, K. et al. Longitudinal study of normal cognition in Parkinson disease. Neurology 85, 1276–1282 (2015).
Pedersen, K. F., Larsen, J. P., Tysnes, O. B. & Alves, G. Prognosis of mild cognitive impairment in early Parkinson disease: the Norwegian ParkWest study. JAMA Neurol. 70, 580–586 (2013).
Williams-Gray, C. H. et al. The CamPaIGN study of Parkinson's disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 84, 1258–1264 (2013).
Aarsland, D., Andersen, K., Larsen, J. P., Lolk, A. & Kragh-Sorensen, P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch. Neurol. 60, 387–392 (2003).
Yarnall, A. J. et al. Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology 82, 308–316 (2014).
Aarsland, D. et al. Mild cognitive impairment in Parkinson disease: a multicenter pooled analysis. Neurology 75, 1062–1069 (2010).
Foltynie, T., Brayne, C. E., Robbins, T. W. & Barker, R. A. The cognitive ability of an incident cohort of Parkinson's patients in the UK. The CamPaIGN study. Brain 127, 550–560 (2004).
Wood, K. et al. Different PD-MCI criteria and risk of dementia in Parkinson’s disease: 4-year longitudinal study. npj Parkinsons Dis. 2, 15027 (2016).
Biundo, R. et al. Diagnostic and screening power of neuropsychological testing in detecting mild cognitive impairment in Parkinson's disease. J. Neural Transm. (Vienna) 120, 627–633 (2013).
Petersen, R. C. et al. Mild cognitive impairment: clinical characterization and outcome. Arch. Neurol. 56, 303–308 (1999).
Winblad, B. et al. Mild cognitive impairment--beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 256, 240–246 (2004).
Goldman, J. G., Weis, H., Stebbins, G., Bernard, B. & Goetz, C. G. Clinical differences among mild cognitive impairment subtypes in Parkinson's disease. Mov. Disord. 27, 1129–1136 (2012).
Williams-Gray, C. H., Foltynie, T., Brayne, C. E., Robbins, T. W. & Barker, R. A. Evolution of cognitive dysfunction in an incident Parkinson's disease cohort. Brain 130, 1787–1798 (2007).
Troster, A. I. Neuropsychological characteristics of dementia with Lewy bodies and Parkinson's disease with dementia: differentiation, early detection, and implications for ‘mild cognitive impairment’ and biomarkers. Neuropsychol. Rev. 18, 103–119 (2008).
Troster, A. I. A precis of recent advances in the neuropsychology of mild cognitive impairment(s) in Parkinson's disease and a proposal of preliminary research criteria. J. Int. Neuropsychol. Soc. 17, 393–406 (2011).
Pagonabarraga, J. et al. A prospective study of delusional misidentification syndromes in Parkinson's disease with dementia. Mov. Disord. 23, 443–448 (2008).
Biundo, R. et al. Anatomical correlates of cognitive functions in early Parkinson's disease patients. PLoS One 8, e64222 (2013).
Aarsland, D., Muniz, G. & Matthews, F. Nonlinear decline of mini-mental state examination in Parkinson's disease. Mov. Disord. 26, 334–337 (2011).
Rektorova, I. et al. Grey matter changes in cognitively impaired Parkinson's disease patients. PLoS One 9, e85595 (2014).
Weintraub, D. et al. Alzheimer's disease pattern of brain atrophy predicts cognitive decline in Parkinson's disease. Brain 135, 170–180 (2012).
Melzer, T. R. et al. Grey matter atrophy in cognitively impaired Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 83, 188–194 (2012).
Lee, J. E. et al. Exploratory analysis of neuropsychological and neuroanatomical correlates of progressive mild cognitive impairment in Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 85, 7–16 (2014).
Hattori, T. et al. Cognitive status correlates with white matter alteration in Parkinson's disease. Hum. Brain Mapp. 33, 727–739 (2012).
Pagonabarraga, J. et al. Pattern of regional cortical thinning associated with cognitive deterioration in Parkinson's disease. PLoS One 8, e54980 (2013).
Pereira, J. B. et al. Initial cognitive decline is associated with cortical thinning in early Parkinson disease. Neurology 82, 2017–2025 (2014).
Hanganu, A. et al. Mild cognitive impairment is linked with faster rate of cortical thinning in patients with Parkinson's disease longitudinally. Brain 137, 1120–1129 (2014).
Ibarretxe-Bilbao, N. et al. Progression of cortical thinning in early Parkinson's disease. Mov. Disord. 27, 1746–1753 (2012).
Mak, E. et al. Baseline and longitudinal grey matter changes in newly diagnosed Parkinson's disease: ICICLE-PD study. Brain 138, 2974–2986 (2015).
Theilmann, R. J. et al. White-matter changes correlate with cognitive functioning in Parkinson's disease. Front. Neurol. 4, 37 (2013).
Zheng, Z. et al. correlates of distinct cognitive impairments in Parkinson's disease. Hum. Brain Mapp. 35, 1325–1333 (2014).
Melzer, T. R. et al. White matter microstructure deteriorates across cognitive stages in Parkinson disease. Neurology 80, 1841–1849 (2013).
Agosta, F. et al. Mild cognitive impairment in Parkinson's disease is associated with a distributed pattern of brain white matter damage. Hum. Brain Mapp. 35, 1921–1929 (2014).
Duncan, G. W. et al. Gray and white matter imaging: a biomarker for cognitive impairment in early Parkinson's disease? Mov Disord 31, 103–110 (2016).
Carlesimo, G. A. et al. Hippocampal abnormalities and memory deficits in Parkinson disease: a multimodal imaging study. Neurology 78, 1939–1945 (2012).
Gorges, M. et al. To rise and to fall: functional connectivity in cognitively normal and cognitively impaired patients with Parkinson's disease. Neurobiol. Aging 36, 1727–1735 (2015).
Baggio, H. C. et al. Cognitive impairment and resting-state network connectivity in Parkinson's disease. Hum. Brain Mapp. 36, 199–212 (2015).
Amboni, M. et al. Resting-state functional connectivity associated with mild cognitive impairment in Parkinson's disease. J. Neurol. 262, 425–434 (2015).
Tessitore, A. et al. Default-mode network connectivity in cognitively unimpaired patients with Parkinson disease. Neurology 79, 2226–2232 (2012).
Olde Dubbelink, K. T. et al. Functional connectivity and cognitive decline over 3 years in Parkinson disease. Neurology 83, 2046–2053 (2014).
Chen, B., Fan, G. G., Liu, H. & Wang, S. Changes in anatomical and functional connectivity of Parkinson's disease patients according to cognitive status. Eur. J. Radiol. 84, 1318–1324 (2015).
Ravina, B. et al. Dopamine transporter imaging is associated with long-term outcomes in Parkinson's disease. Mov. Disord. 27, 1392–1397 (2012).
Christopher, L. et al. Combined insular and striatal dopamine dysfunction are associated with executive deficits in Parkinson's disease with mild cognitive impairment. Brain 137, 565–575 (2014).
Klein, J. C. et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology 74, 885–892 (2010).
Acosta-Cabronero, J., Williams, G. B., Pengas, G. & Nestor, P. J. Absolute diffusivities define the landscape of white matter degeneration in Alzheimer's disease. Brain 133, 529–539 (2010).
Picconi, B., Piccoli, G. & Calabresi, P. Synaptic dysfunction in Parkinson's disease. Adv. Exp. Med. Biol. 970, 553–572 (2012).
Greicius, M. D., Krasnow, B., Reiss, A. L. & Menon, V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc. Natl Acad. Sci. USA 100, 253–258 (2003).
Marcus, E. Credibility and reproducibility. Chem. Biol. 22, 3–4 (2015).
Spreng, R. N., Sepulcre, J., Turner, G. R., Stevens, W. D. & Schacter, D. L. Intrinsic architecture underlying the relations among the default, dorsal attention, and frontoparietal control networks of the human brain. J. Cogn. Neurosci. 25, 74–86 (2013).
Fox, M. D., Corbetta, M., Snyder, A. Z., Vincent, J. L. & Raichle, M. E. Spontaneous neuronal activity distinguishes human dorsal and ventral attention systems. Proc. Natl Acad. Sci. USA 103, 10046–10051 (2006).
Spreng, R. N., Stevens, W. D., Chamberlain, J. P., Gilmore, A. W. & Schacter, D. L. Default network activity, coupled with the frontoparietal control network, supports goal-directed cognition. Neuroimage 53, 303–317 (2010).
Seeley, W. W. et al. Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci 27, 2349–2356 (2007).
Griffanti, L. et al. Individual thresholding of voxel-based functional connectivity maps. Estimation of random errors by means of surrogate time series. Methods Inf. Med. 54, 227–231 (2015).
van den Heuvel, M. P., Stam, C. J., Kahn, R. S. & Hulshoff Pol, H. E. Efficiency of functional brain networks and intellectual performance. J. Neurosci. 29, 7619–7624 (2009).
Menon, V. Large-scale brain networks and psychopathology: a unifying triple network model. Trends Cogn. Sci. 15, 483–506 (2011).
Putcha, D., Ross, R. S., Cronin-Golomb, A., Janes, A. C. & Stern, C. E. Altered intrinsic functional coupling between core neurocognitive networks in Parkinson's disease. Neuroimage Clin. 7, 449–455 (2015).
Sridharan, D., Levitin, D. J. & Menon, V. A critical role for the right fronto-insular cortex in switching between central-executive and default-mode networks. Proc. Natl Acad. Sci. USA 105, 12569–12574 (2008).
Anticevic, A. et al. The role of default network deactivation in cognition and disease. Trends Cogn. Sci. 16, 584–592 (2012).
Menon, V. & Uddin, L. Q. Saliency, switching, attention and control: a network model of insula function. Brain Struct. Funct. 214, 655–667 (2010).
Dang, L. C., O'Neil, J. P. & Jagust, W. J. Dopamine supports coupling of attention-related networks. J. Neurosci. 32, 9582–9587 (2012).
Criaud, M. et al. Contribution of insula in Parkinson's disease: a quantitative meta-analysis study. Hum. Brain Mapp. 37, 1375–1392 (2016).
Bohnen, N. I. et al. Frequency of cholinergic and caudate nucleus dopaminergic deficits across the predemented cognitive spectrum of Parkinson disease and evidence of interaction effects. JAMA Neurol. 72, 194–200 (2015).
Merdes, A. R. et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 60, 1586–1590 (2003).
Aarsland, D., Perry, E., Perry, R., Larsen, J. & Ballard, C. Duration of Parkinsonism prior to dementia is associated with a different pattern of<br/>neuropathological and neurochemical substrates in DLB and PDD (Poster Sessions: Clinical Science). Mov. Disord. 21, S96 (2006).
Biundo, R. et al. MMSE and MoCA in Parkinson's disease and dementia with Lewy bodies: a multicenter 1-year follow-up study. J. Neural Transm. (Vienna) (2016).
Author information
Authors and Affiliations
Contributions
RB: wrote the manuscript. LW: wrote a section of the manuscript and review the manuscript. AA: edited the final manuscript. All authors contributed intellectual material.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Biundo, R., Weis, L. & Antonini, A. Cognitive decline in Parkinson’s disease: the complex picture. npj Parkinson's Disease 2, 16018 (2016). https://doi.org/10.1038/npjparkd.2016.18
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/npjparkd.2016.18
This article is cited by
-
Clinical biomarkers for Lewy body diseases
Cell & Bioscience (2023)
-
Morphological basis of Parkinson disease-associated cognitive impairment: an update
Journal of Neural Transmission (2022)
-
Impact of social and mobility restrictions in Parkinson’s disease during COVID-19 lockdown
BMC Neurology (2021)
-
TOMM40 ‘523’ poly-T repeat length is a determinant of longitudinal cognitive decline in Parkinson’s disease
npj Parkinson's Disease (2021)
-
Multivariate prediction of dementia in Parkinson’s disease
npj Parkinson's Disease (2020)
