
Abstract
Diabetes mellitus and periodontal disease are chronic diseases affecting a large number of populations worldwide. Changed bone metabolism is one of the important long-term complications associated with diabetes mellitus. Alveolar bone loss is one of the main outcomes of periodontitis, and diabetes is among the primary risk factors for periodontal disease. In this review, we summarise the adverse effects of diabetes on the periodontium in periodontitis subjects, focusing on alveolar bone loss. Bone remodelling begins with osteoclasts resorbing bone, followed by new bone formation by osteoblasts in the resorption lacunae. Therefore, we discuss the potential mechanism of diabetes-enhanced bone loss in relation to osteoblasts and osteoclasts.
Similar content being viewed by others

Diabetes: introduction
Diabetes mellitus is a heterogeneous group of disorders and is characterized by high blood glucose levels.1 Type 1 diabetes mellitus (T1DM) results from an absolute deficiency of insulin, which is most commonly due to auto-immunological destruction of the insulin-producing pancreatic β cells but which can be caused by other etiologies. In type 2 diabetes mellitus (T2DM), muscle, fat and other cells become resistant to the actions of insulin. This results in the activation of a compensatory mechanism that induces β cells to secrete more insulin. T2DM occurs when the compensatory increase in insulin is insufficient to maintain blood glucose levels within a normal physiological range.2,3 By 2025, 300 million people are projected to be afflicted with diabetes worldwide, with a prevalence of 6.4%.4,5 The countries with the most people suffering from diabetes by the year 2025 are predicted to be India, China and the United States. T1DM represents 5%–10% of the total number of diabetes cases worldwide6 and is the main type of diabetes in youth, representing 85% or more of all diabetes cases in individuals younger than 20 years of age worldwide.7 On average, males and females are equally affected with T1DM in young populations.8 T2DM accounts for 90% of diabetes cases globally.4 This disorder has traditionally been considered a metabolic disorder of adults; however, it has recently become more common in young adults, adolescents and occasionally, in children.9
Pathogenic mechanisms of diabetes
T1DM is a polygenic autoimmune disease that is characterized by the destruction of insulin-secreting pancreatic beta cells.10 T1DM typically occurs as a consequence of a breakdown in immune regulation, resulting in an expansion of auto-reactive CD4+ and CD8+ T cells and auto-antibody-producing B lymphocytes and activation of the innate immune system, which collaborates to destroy insulin-producing beta cells.11 In an animal model, CD11c+ dendritic cells and ER-MP23+ macrophages are the first cells to infiltrate the pancreas of non-obese diabetic mice, at approximately 3 weeks of age. At the same time, potentially pathogenic T cells can be detected surrounding the islet, resulting in peri-insulitis.12 Genetic susceptibility and environmental triggers are thought to contribute to the development of T1DM.13
T2DM is a metabolic disorder that is characterized by hyperglycemia and altered lipid metabolism, which is caused by the inability of islet β cells to secrete adequate insulin in response to varying degrees of insulin resistance caused by over-nutrition, inactivity or obesity. Metabolic defects that contribute to the development of T2DM include an inability of islet β cells to compensate for high glucose levels that are associated with excess food intake, increased glucagon secretion and reduced incretin response, impaired expansion of subcutaneous adipose tissue, hypoadiponectinaemia, inflammation of adipose tissue, increased endogenous glucose production and the development of peripheral insulin resistance.14 Chronic caloric excess is the primary pathogenic event that drives the development of type 2 diabetes in genetically and epigenetically susceptible individuals.15,16
Pathogenic changes in both T1DM and T2DM
Hyperglycemia
Hyperglycemia is due to impaired insulin secretion in T1DM and insulin resistance in T2DM. Beta cell destruction in T1DM and inadequate expression of glucose transporter 2 in T2DM are thought to contribute to hyperglycemia.17 Several pathways are thought to exhibit increased activity under hyperglycemic conditions and to contribute to oxidative stress via the polyol pathway,18 the hexosamine pathway 19,20,21 and activator of protein kinase C.22 Hyperglycemia also leads to greater activation of the pro-inflammatory transcription factor, nuclear factor-kappa B (NF-κB), by protein kinase C in vitro.19 Hyperglycemia also results in the oxidation of sorbitol by NAD+, thereby increasing the cytosolic NADH:NAD+ ratio and consequently inhibiting glyceraldehyde-3-phosphate dehydrogenase activation.
Advanced glycation end products
Advanced glycation end products (AGEs) are formed by the non-enzymatic reaction of glucose and other glycating compounds that are derived from glucose and increased fatty acid oxidation. Intracellular hyperglycemia is the primary initiating event in the formation of both intracellular and extracellular AGEs.23 AGEs are derived from the intracellular auto-oxidation of glucose to glyoxal,24 decomposition of the Amadori product (glucose-derived 1-amino-1-deoxyfrutose lysine adducts) and fragmentation of glyceraldehade-3-phosphate and dihydroxyacetone phosphate to methylglyoxal.25 These reactive intracellular dicarbonyls (glyoxal, methylglyoxal and 3-deoxyglucosone) react with the amino groups of intracellular and extracellular proteins to form AGEs. Intracellular proteins that are modified by AGEs exhibit altered function. Extracellular matrix components that have been modified by AGE precursors interact abnormally with other matrix proteins and their receptors on cells. Several AGE receptors are linked to increased inflammation, including receptor for AGE (RAGE). Proteins can be structurally modified by glycosylation, thereby affecting their function. Alternatively, AGE binding to AGE receptors can induce the production of reactive oxygen species, the production of inflammatory cytokines such as tumour necrosis-alpha (TNF-α), and the activation of NF-κB.26
Lipotoxicity
Due to the presence of long-chain free fatty acids in the plasma, lipotoxicity is often increased in states of insulin resistance, thereby impairing β-cell secretory function 27,28 and contributing to β-cell apoptosis 29,30 and insulin resistance.31 Muscle cells and hepatocytes are negatively affected by excessive amounts of fatty acids, which cause increased ceramide accumulation, activate inflammatory pathways, and increase the release of reactive oxygen species (ROS) and enhance apoptosis.32
Oxidative stress
Oxidative stress is central to the development of insulin resistance and diabetic complications.33,34 Oxidative stress plays a critical role in diabetic complications. Hyperglycemia leads to the overproduction of superoxides in mitochondria. This increase in superoxide production activates several pathways that contribute to diabetic complications, including polyol pathway flux, increased AGE formation and RAGE expression, and activation of protein kinase C and the hexosamine pathway.18 Inflammation induced by increased intracellular ROS also contributes to diabetic complications.35 After ROS, are created, they deplete cellular antioxidant defences, rendering the affected cells and tissues more susceptible to oxidative damage.36 It has been shown that ROS not only play a role in the destruction of cells and tissues but also function as intracellular second messengers that regulate signal transduction cascades and gene expression. Oxidative stress can also induce the activation of multiple serine kinases, which impair the capacity of insulin to stimulate protein kinase B activation and glucose transport. NF-κB, p38 MAPK and the JNK/SAPK pathway are sensitive to oxidative stress, which is linked to impaired insulin action and the development of the late diabetic complications.33
Immune response
Neutrophils play a crucial role in several autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis. Some studies have noted the involvement of neutrophils in T1DM; a mild neutrophil reduction has been associated with T1DM subjects;37,38 however, neutrophil counts in T2DM patients are normal.37 The reduction in circulating neutrophils that is observed in T1DM might be due to impaired neutrophil differentiation and output from bone marrow, increased neutrophil apoptosis or anti-neutrophil-specific antibodies, and increased recruitment into tissues.39 The functional alteration of mononuclear phagocytes has also been reported in diabetic subjects, including altered superoxide (O2−) production, defective chemotaxis, and phagocytosis.40 A study has shown that infiltrating monocytes in T1DM subjects spontaneously secrete pro-inflammatory cytokines, which are known to induce and expand Th17 cells.41 Evidence also shows that the classically activated macrophages initiate insulitis and β-cell death in T1DM subjects and play a role in insulin resistance in T2DM by triggering an inflammatory response. In contrast, alternatively activated macrophages exert a protective effect in DM by attenuating tissue inflammation.42
Pro-inflammatory factors
Adipose tissue appears to be a major site for inflammatory mediator production as a result of cross-talk between adipose cells, macrophages, and other immune cells that infiltrate the expanding adipose tissue.43 Inflammatory mediators might play a dual role in T2DM, contributing to hyperglycemia-induced insulin resistance and contributing to diabetic complications.44 Pro-inflammatory factors, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IL-18, are reportedly increased in diabetes mellitus and contribute to insulin resistance by both JNK and the IKKβ/NF-κB pathway.45,46 Enhanced production of inflammatory cytokines is thought to contribute to insulin resistance and the destruction of beta cells in the pancreas and is thought to be a major factor in the development of diabetic complications.47,48
Anti-inflammatory factors
The role of pro-inflammatory cytokines in the destruction of pancreatic β cells and the development of T2DM has been investigated; however, our knowledge about anti-inflammatory proteins is rather limited, and an imbalance between pro- and anti-inflammatory cytokines might be essential for the development of DM.49,50 IL-1 receptor antagonist is a naturally occurring anti-inflammatory antagonist of the IL-1 family of pro-inflammatory cytokines.51 Evidence shows that blocking IL-1β signals reduces the expression of inflammatory cytokines, and IL-1 receptor antagonist improves glycemic control and counteracts β-cell destruction.52 Acting as a hormone with anti-inflammatory insulin-sensitizing properties, adiponectin reportedly associates with a decrease in the risk of T2DM.53 However, increased circulating levels of adiponectin are found in T1DM patients, this might be explained by the dependence of T2DM on insulin resistance.50
Complications of diabetes
T1DM and T2DM have many possible long-term complications. Epidemiological studies indicate that the severity of diabetic complications is generally proportional to the degree of hyperglycemia.54
Macrovascular complications
The injurious effects of diabetes on the vascular system are traditionally divided into macrovasular and microvascular complications.55 Evidence suggests that diabetic patients are three times more likely to suffer cardiovascular events than are non-diabetic subjects.56 Cardiovascular disease is the most common cause of death in diabetic patients.57 The central pathological mechanism in macrovascular disease is atherosclerosis. Atherosclerosis is thought to result from chronic inflammation of the arterial wall in the peripheral or coronary vascular system. One mechanism is the stimulation of lipid oxidation of low-density lipoprotein in diabetes mellitus, which accumulates in the endothelial wall of arteries.
Microvascular complications
High serum glucose levels affect endothelial cells that line blood vessels and cause the basement membrane to become thicker and less effective.58 Microvascular complications are a significant part of diabetic retinopathy, nephropathy and neuropathy.59 Diabetes-enhanced TNF induces the loss of microvascular endothelial cells and pericytes by activating the transcription factor FOXO1.60,61
Retinopathy
Diabetic retinopathy is clinically classified into non-proliferative and proliferative disease stages. Only intraretinal microvascular changes are present in non-proliferative diabetic retinopathy, whereas new blood vessels are formed on the retina or on the optic disk in proliferative diabetic retinopathy.62 Diabetic retinopathy is characterized by several common and unique features, including thickening of the vascular basement membrane, pericyte and endothelial cell death, microaneurysms, vascular occlusion and pathological neovascularisation, which advance to retinal haemorrhage, retinal detachment, and vision loss.63 TNF has been shown to play an important role in stimulating the early events of diabetic retinopathy.60
Neuropathy
Diabetic neuropathy is characterized by progressive neuronal loss, demyelination, and impaired nerve regeneration, with ultimate nerve fibre dysfunction.62 Diabetic neuropathy can affect sensory, motor and autonomic nerve fibres in any part of the body.64 Although diabetic neuropathy has been widely studied for over 20 years, the pathogenesis of this disease remains unclear; diabetic neuropathy is thought to result from the diabetic microvascular injury of small blood vessels that supply nerves, as well as oxidative injury, AGEs and insulin deficiency.65
Nephropathy
Hyperglycemia induces cellular changes in various kidney cell types. Nephropathy is a progressive kidney disease that is caused by the angiopathy of capillaries in the kidney glomeruli and is characterized by glomerular hypertrophy, thickening of basement, tubular and glomerular membranes and the accumulation of extracellular matrix in these membranes; these changes finally cause tubulointerstitial and glomerular fibrosis and sclerosis.66 AGEs, hyperglycemia and vascular inflammation are thought to cause these pathogenic changes.67,68
Diabetes-related bone diseases
In addition to the complications mentioned above, diabetes affect bone metabolism. A considerable amount of evidence has accumulated indicating that metabolic and endocrine alterations caused by diabetes affect bone quantity and quality over the last decades of life.69
Increased fracture risk
T1DM and T2DM are associated with higher fracture risk. A large case control study in Denmark reported a relative risk for any site fracture of 1.9.70 The relative risk of hip fracture is estimated as 6.9 in T1DM patients and as 1.38 in T2DM patients.71 Women with T2DM have a threefold higher risk of vertebral fracture compared to women who are non-diabetic.72 In the women's health initiative study, the risk of proximal humerus, foot, and ankle fractures was found to be higher among women with type 2 diabetes than among healthy controls.73
Changed bone mineral density
Bone mineral density (BMD) is reduced in T1DM.72,74 Decreased BMD and diabetic vascular complications are associated with an increased risk of fractures. In T2DM, BMD is equal or increased according a meta-analysis,71 but the fracture risk is increased despite this increase in BMD.75 The paradoxical increase in fracture rate in patients with T2DM with increased BMD might result from an increased rate of falling.76 In addition, decreased bone formation with decreased bone quality is thought to contribute to a higher fracture risk in T2DM. Rodent models also support the hypothesis that diabetic bone has reduced mechanical strength, regardless of bone density.77
Lower bone turnover rate
Decreased bone turnover rate might explain the increased bone fragility in patients with diabetes. Older bone is not replaced by new bone with decreased bone turnover rate, thus decreasing bone mechanical strength. This is reflected in studies indicating that diabetic patients exhibit decreased osteoblast activity.78 Low bone turnover accompanied by reduced mineral content was found in T1DM models.79 Leptin receptor-deficient mice, a T2DM model, also exhibit decreased bone turnover.80,81 The impact of diabetes on bone resorption has yielded contradictory findings, with some studies indicating increased osteoclast activity under perturbation.82,83
Delayed fracture healing
Fracture healing is delayed in diabetic patients. A study of 5 966 cases of hip fracture in patients with diabetes reported that these patients required longer in-hospital stays.84 Moreover, diabetic patients undergoing ankle fusing procedures exhibit an increased incidence of non-union outcomes (from 27% to 38%).85 In diabetic mice that were fed a high fat diet, fracture calluses were significantly smaller and had markedly increased adiposity.86 In streptozotocin-induced diabetic mice, fracture calluses exhibited reduced bone formation, which was associated with premature cartilage resorption.87
Potential mechanisms by which diabetes affects bone
Bone formation and resorption affect bone, and diabetes mellitus affects both bone formation and resorption.
Effect of diabetes on osteoblasts
Diabetes has a significant effect on osteoblasts. It has consistently been noted that diabetes causes a reduction in the number of bone-forming cells.88 One of the mechanisms through which diabetes affects osteoblasts is increased apoptosis. For example, AGEs induce osteoblast apoptosis through the MAP kinase pathway.89 Diabetes also interferes with bone formation by reducing the expression of transcription factors that regulate osteoblast differentiation.90 In rat models of T1DM and T2DM, osteoblasts exhibit lower alkaline phosphatase activity and mineralized matrix formation.91,92 When AGEs are applied to wounds in normal animals, the rate of healing is reduced by half, suggesting that AGEs, which are present at higher levels in diabetes, contribute to impaired bone healing.93 In addition, the receptor for AGEs, RAGE, is expressed at higher levels in osteoblasts in diabetic conditions, thus rendering diabetic animals even more sensitive to the effects of AGEs.93
Inflammation also has a significant effect on bone.94 Elevated levels of inflammatory mediators, particularly TNF, are among the striking features of diabetes.95 Diabetic animals exhibit higher levels of TNF in bone, and enhanced TNF levels are associated with reduced bone healing. Thus, diabetes-enhanced TNF contributes to an increase in bone cell apoptosis, which reduces osseous healing.96 Moreover, diabetes causes a more than two-fold induction of more than 70 genes that directly or indirectly regulate apoptosis during inflammation-induced bone injury and that significantly enhance caspase-8, -9 and -3 activity.97 A significant effect on bone, which was induced by cell death in diabetic animals, was shown by treating mice with a pancaspase inhibitor. Treatment with this inhibitor increased the number of bone-lining cells and enhanced new bone formation.97 Profiling of mRNA expression during diabetic fracture indicates that gene sets that are related to apoptosis are significantly upregulated.98 The increased expression of apoptotic genes is associated with elevated TNF-α levels, increased chondrocyte apoptosis and increased activity of the pro-apoptotic transcription factor, FOXO1. Inhibition of TNF significantly reduces each of these effects in diabetic fracture healing but has little effect in normoglycemic mice.98
Mesenchymal stem cells represent a precursor pool of osteoblasts, which are bone-forming cells. Inflammation, which is elevated in diabetic bone healing,96 has a significant effect on reducing mesenchymal stem cell (MSC) differentiation.99 Inflammation affects MSC through the induction of NF-κB activation. Increased NF-kB activity interferes with wnt-stimulated MSC differentiation by increasing beta-catenin degradation.100 Moreover, TNF suppresses Osx promoter activation,101 thus interfering with the differentiation of MSCs to osteoblasts because osterix is required during the early steps of differentiation. AGEs also inhibit MSC differentiation. One mechanism by which this occurs involves the upregulation of ROS by AGEs in MSCs, leading to a decrease in MSC differentiation.102,103 In human mesenchymal stem cells and mouse stromal ST2 cells, AGEs suppress the osteogenic differentiation of both cell types by increasing TGF-β expression.104 In a T2DM mouse model, the diabetic mouse has fewer MSCs, and these MSCs exhibit poor ability to target the injury site.105 In a T1DM rat, more numerous apoptotic cells were present in hyperglycemic bone marrow, and the size of the osteoprogenitor pool was significantly reduced.92 Thus, elevated levels of TNF and the effects of AGEs might interfere with the production of osteoblasts that are formed by inhibiting the differentiation of MSC to osteoblasts.
Effect of diabetes on osteoclasts
Human studies of diabetes mellitus generally indicate that osteoclastogenesis is enhanced. Humans with T2DM exhibit increased circulating levels of tartrate-resistant acid phosphatase, which is indicative of increased osteoclast activity.82 In patients with T1DM or T2DM, poor glycemic control leads to increased bone resorption and bone loss.78 In patients with T2DM, levels of the bone resorption marker tartrate-resistant acid phosphatase are increased in serum, indicating increased osteoclastic function, which might be due to the diabetic polyol pathway.106 However, some studies indicate that bone resorption markers are decreased in individuals with T2DM.107
Animal studies generally indicate that osteoclast activity is increased in diabetic patients.108,109 In streptozotocin-induced T1DM rats, cathepsin K expression is elevated compared to a control group, indicating increased osteoclast activity.110 In T2DM rats, osteoclastic bone resorption is enhanced compared to normoglycemic controls.111 In diabetic mice, TNF-α, macrophage-colony stimulating factor, receptor activator of nuclear factor kappa-B ligand (RANKL) and vascular endothelial growth factor-A are upregulated; this upregulation can directly promote osteoclast differentiation and activation.87,112 Patients with T2DM exhibit elevated mitochondrial ROS levels, which promote RANKL-mediated osteoclast differentiation and function.113 Increased fatty acid levels in patients with diabetes mellitus can induce osteoclastogenesis by TNF-α.114 T2DM mice exhibited enhanced osteoclast formation in response to M-CSF and RANKL.82In vitro studies suggest that hyperglycemia predisposes patients to increased osteoclast formation.115 AGEs might also increase osteoclast activity.116 Mice that lack the receptor for AGE, RAGE, exhibit increased bone mass and decreased osteoclast numbers compared to wild-type mice,117 supporting the concept that AGEs contribute to osteoclast formation in patients with diabetes.
Periodontal disease: introduction
Periodontitis is one of the most widespread oral diseases and is characterized by the loss of connective tissues within the periodontium and the destruction of alveolar bone support.118,119 Severe periodontitis, which can result in tooth loss, is found in 5%–20% of most adult populations worldwide. The latest data from the 2009 and 2010 National Health and Nutrition Examination Survey cycle estimates that over 47% American adults have had periodontitis.120 An epidemiology study shows that almost 25% of adults in Australia aged 35–54 years have moderate or severe periodontitis, and 34% of 30- to 39-year-old adults living in Pomerania have periodontitis.121 Children and adolescents can have any of the several forms of periodontitis, such as aggressive periodontitis, chronic periodontitis and periodontitis as a manifestation of systemic diseases.122 However, estimates of the global prevalence of periodontitis differ based on the distribution of the disease and the methodologies used to assess it.123
Pathogenic mechanisms of periodontal disease
Microorganisms
The chronic inflammatory condition of periodontitis is induced by pathogenic biofilms or dental plaque, which accumulates on the tooth surface. Over 500 bacterial species have been detected in periodontal plaque; however, the composition of the causative bacterial species is still under debate.123,124,125,126 ‘Red complex’ Gram-negative bacteria, comprising Porphyromonas gingivalis, Tannerella forsythia and Treponema denticola, have been proposed as the primary aetiological agents of periodontitis.127 Recent studies have identified Filifactoralocis and many species of Spirochetes as being closely linked to periodontitis.128 As the best characterized periopathogen, P. gingivalis is a minor constituent of the total microbiota but can trigger changes in the amount and composition of the oral commensal microbiota that can disrupt homeostasis and cause inflammatory periodontal bone loss.129 Gram-positive bacterial and oral commensal bacteria might also play a significant role in the development of periodontitis 128,130,131.
Modern high-throughput techniques indicate that the subgingival microbial profiles from periodontitis patients differ according to age, pocket depth, gender and race.128,132,133 Interestingly, bacteria community diversity has been shown to increase with a history of periodontitis, which runs counter to most bacterial infectious processes, which are generally associated with decreased diversity.128
Host response
Although bacteria are necessary for periodontal disease to occur, a susceptible host is also needed.123 The inflammatory process occurring in periodontitis is characterized by the infiltration of leukocytes, which limit the level of bacterial invasion and can be harmful to the periodontal tissue.134 Periodontal ligament and bone destruction is thought to be caused by a disruption of the homeostatic balance between the host response and bacteria, which causes inflammation in close proximity to bone.129,134,135 The process is thought to involve the host immune response to bacterial infection through the stimulation of osteoclastogenic factor production by immune cells, which then help to cause the periodontitis-related bone loss. Our laboratory has recently shown that the production of factors by osteoblasts and osteocytes also contributes to osteoclast formation and activity in periodontal disease (unpublished data). Evidence that the host response plays a critical role has also shown by studies in which treatment with a prostaglandin inhibitor reduced periodontitis-related bone loss136 and studies involving the inhibition of inflammatory cytokines, such as IL-1 and TNF.137,138 Thus, periodontitis is a complex disease in which multiple causal risk factors play simultaneous and interactive roles; these risk factors include the immune-inflammatory status and genetic background of individuals, and the presence of environmental stressors and/or systemic diseases, such as diabetes.139,140,141
Effects of diabetes on periodontitis
Diabetes and chronic periodontitis are chronic diseases that have long been considered to be biologically linked.142,143 In fact, diabetes is one of the primary risk factors for periodontitis.144,145 Cross-sectional and longitudinal studies identified that the risk of periodontitis is approximately 3–4 times higher in people with diabetes than in non-diabetic subjects.144 Periodontitis was found in 57.9% of T1DM patients and in 15.0% in controls without diabetes.146 In another study on the periodontal status in children and adolescents with T1DM, a prevalence of 20.8% of gingivitis and 5.9% of periodontitis was observed.147 Patients with T2DM were also at higher risk of having severe forms of periodontitis compared with non-diabetic subjects.148 A study in African Americans found that 70.6% of patients with T2DM had moderate periodontitis, and 28.5% had a severe form of the disease; this value is significantly higher than the prevalence of 10.6% among control subjects without diabetes.149 A direct relationship exists between the level of glucose control and the severity of periodontitis.146,150 The odds ratios of T2DM with periodontal destruction in comparison to non-diabetics are 1.97, 2.10 and 2.42 in well, moderately and poorly controlled diabetics, respectively.151
Effects of diabetes on periodontium tissues
Gingiva/gingivitis
Periodontitis is preceded by various stages of gingival inflammation, which are referred to as gingivitis. The prevalence of gingivitis in children and adolescents with T1DM is nearly twice that observed in children and adolescents without diabetes.152 Evidence shows that the gingival index was 1.54 in a 5- to 9-year-old diabetic group and 1.14 in the control group; however, in the corresponding 10- to 14-year-old group, the gingival index was 1.98 in diabetic subjects and 1.17 in control subjects.153 Furthermore, gingival bleeding index is significantly correlated with age and the level of glycosylated haemoglobin.154 Similarly, the rates of gingival inflammation in adults with T2DM are higher than those in adults without diabetes. Nearly 64% of patients with T2DM have gingivitis; however, only 50% of subjects without diabetes have the disease.152 The degree of metabolic control of diabetes is an important factor in the development and progression of gingivitis; good control significantly reduces the prevalence of gingivitis.155,156 In animal models, diabetes leads to the increased production of TNF in the epithelium and connective tissue.157 Periodontal infection causes an increase in epithelial cell and connective tissue fibroblast apoptosis, which is significantly enhanced by diabetes through a caspase-3-dependent mechanism.157 This is significant because increased diabetes-enhanced inflammation and apoptosis are thought to negatively impact the gingiva by causing a loss of epithelial barrier function and inhibiting repair processes.158,159 Animal studies have demonstrated that high levels of TNF-α can stimulate the expression of pro-apoptotic genes, which induce apoptosis.96,160 In vivo studies have demonstrated that the inhibition of TNF-α reduces connective tissue cell apoptosis.161
Periodontal ligament/loss of attachment
Periodontitis involves the loss of tooth-supporting structures comprising connective tissue attachment and bone. A statistically significant association is observed between diabetes and loss of attachment, and the level of glycemic control is an important determinant of this relationship.146,162 The odds ratio of T1DM patients with attachment loss is 3.84 compared with non-diabetic subjects.163 In addition, more than 25% of T1DM patients with poor metabolic control exhibit sites with clinical attachment loss of ≥5 mm, compared with 10% of subjects with good metabolic control.152 Subjects with T2DM also exhibit a significantly higher number of sites with clinical attachment loss of ≥3 mm as well as loss of ≥5 mm. Furthermore, the prevalence of sites with moderate to severe attachment loss is proportional to the duration of diabetes.150,164
Alveolar bone/loss of bone
Diabetes potentiates the severity of periodontitis and accelerates bone resorption. The percentage of sites with bone loss in poorly controlled T1DM individuals is 44% compared to 28% and 24% in well-controlled and non-diabetic subjects, respectively.165 Animal studies also demonstrate that alveolar bone loss in rats with STZ-induced T1DM with periodontitis was threefold higher than in normal rats.166,167 The risk and degree of alveolar bone loss is positively correlated with lack of metabolic control.168,169
Mechanisms of diabetes enhanced periodontal bone loss
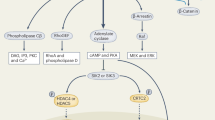
The process of bone remodelling starts with the resorption of bone by osteoclasts, followed by new bone formation by osteoblasts in the resorption lacunae. Under physiological conditions, these two activities are coupled; however, the two processes are uncoupled in pathological processes.118,134 Diabetes affects osteoclast and osteoblasts in the periodontium in different ways, such as by increasing the expression of inflammatory mediators and RANKL/osteoprotegerin (OPG) ratios and by enhancing the levels of AGEs and ROS (Figure 1).

A potential mechanism of diabetes-related alveolar bone loss in periodontal disease. Diabetes increases the RANKL/OPG ratios and enhances the expression level of AGEs, ROS and inflammatory mediators, which induce osteoblast apoptosis and osteoclastogenesis. This cascade of events contributes to both increased bone resorption and reduced reparative bone formation leading to greater alveolar bone loss in periodontal disease that is caused by bacterial pathogens. AGE, advanced glycation end product; IL, interleukin; OPG, osteoprotegerin; PDL, periodontal ligament; RANKL, receptor activator of nuclear factor kappa-B ligand; ROS, reactive oxygen species; TNF, tumor necrosis factor.
Effect of diabetes on osteoclasts in periodontitis
Diabetes has been shown to enhance osteoclast formation in inflamed areas. Rats with T2DM exhibit a two- to four-fold increase in osteoclast number after bacterial infection by oral inoculation of a periodontal pathogen ligature that induces periodontitis compared with control rats.111,157,170 Rats with T1DM and periodontitis also exhibit a two- to four-fold increase in the number of osteoclasts compared to non-diabetic rats with periodontitis.171
A higher degree of inflammation and a more persistent inflammatory response following periodontitis are reported in rats with T1DM and T2DM in response to the same inoculation of periodontal pathogen.172,173 Diabetes impairs the resolution of periodontal inflammation. The importance of resolving inflammation has been demonstrated by treating animals with periodontitis with resolvins174 or by treatment of diabetic animals with TNF inhibitors.160,170 Diabetes-enhanced TNF has been shown to prevent the downregulation of genes that are associated with host defence, apoptosis, cell signalling and activity, and coagulation/homeostasis/complement.175 Similarly, patients with periodontitis and diabetes were found to have significantly higher levels of local inflammatory mediators such as IL-1β, TNF-α and prostaglandin E2, which result in more prolonged osteoclast formation and activity.176 Enhancement of IL-17 and IL-23 in periodontitis in subjects with T1DM and overexpression of IL-1β and IL-6 in patients with T2DM have been reported; and these conditions result in osteoclastogenesis and a prolonged duration of inflammatory responses.177,178 Patients with T2DM and periodontal disease exhibit increased levels of TNF-α and IL-6, which are also associated with increased dyslipidemia and lipid peroxidation.179 These results suggest that there is an important association between T2DM, dyslipidemia and the severity of the local inflammatory response to bacteria.179
The interaction of RANKL with receptors (RANK) on the surface of osteoclasts is one of the most potent inducers of osteoclast formation and activity, and OPG inhibits osteoclast formation binding to RANK and then blocks the activity of RANKL.169,170,180 A number of studies focusing on osteoclastogenesis-related factors have reported an elevated expression of RANKL and TNF in diabetes-associated periodontal tissues.180 Studies with animals suggest that RANK-RANKL/OPG ratios and the level of other inflammatory cytokines, such as TNF, are critical mediators of the enhanced osteoclastogenesis in diabetes with periodontal disease.170,171,181 TNF levels and the RANKL/OPG ratio in periodontitis sites in humans are negatively influenced by poor glycemic control in subjects with diabetes.179,182
Diabetes enhances the formation of AGEs in the periodontium and increases the expression of RAGE.183 Gingiva AGEs are increased in both T1DM- and T2DM-associated periodontitis; however, there is evidence that subjects affected with T1DM presented a significantly higher percentage of AGE-positive cells in the epithelium and fibroblasts than did subjects affected with T2DM.184 Osteoclast-like cells express RAGE, which serves as a positive factor that regulates osteoclast formation.117 AGE accumulation and the interaction of AGEs with RAGE may contribute to osteoclastogenesis via an increased expression of the receptor activator of RANKL and OPG downregulation.185 It has been shown that the AGE-RAGE interaction on monocytes activates transcription factor NF-κB, which alters the phenotype of monocytes/macrophages and results in the increased production of pro-inflammatory cytokines.186
ROS are known to be one of causes of diabetes-related periodontitis. The invading bacteria trigger the release of inflammatory cytokines, leading to increases in the number and activity of neutrophils, which release ROS in periodontitis.187 During bone resorbtion, osteoclasts that contain NADPH-oxidase actively produce superoxide.188 Simultaneously, neutrophils in diabetic patients produce more superoxide than neutrophils from normal subjects.26 The imbalance between ROS production and antioxidant defences result in increased oxidative stress.189 In addition, the formation of AGEs also increases oxidative stress in periodontium tissue. It has been shown that certain ROS (such as superoxide and hydrogen peroxide) activate osteoclasts and promote osteoclast formation.190 A related process, lipid peroxidation, is also associated with increased periodontal disease-T2DM and a greater inflammatory response in periodontal tissues in humans.179,191
Effect of diabetes on osteoblasts in periodontitis
Evidence exists that both diabetes and bacterial infection in periodontitis enhance the apoptosis of osteoblastic cells, thereby reducing osseous coupling.161,192 It is likely that both the innate and adaptive immune responses that are induced by bacterial infection cause osteoblastic cell loss.96,193 Diabetes also increases the loss of periodontal ligament (PDL) cells that is induced by periodontal infection by increasing the apoptosis of these cells.111,194 This loss is significant because the PDL is a rich source of cells that are capable of differentiating into osteoblasts. Studies in diabetic animals indicate that diabetes causes a more than 2-fold induction of genes that regulate the apoptosis of osteoblasts and fibroblasts following bacterial infection and a fivefold increase in osteoblast apoptosis.97,195 The apoptosis of osteoblasts is a significant component of the failure of diabetic animals to form new bone after the induction of periodontal disease, as shown by a significant increase in bone formation when diabetic animals are treated with a specific blocker of apoptosis following periodontal infection.161
Diabetes leads to the upregulation of pro-apoptotic factors for osteoblasts, including advanced TNF-α, AGEs and the formation of ROS, each of which can contribute to apoptosis.161 Animals with T1DM and T2DM exhibit higher levels of TNF-α in response to a bacterial stimulus than normoglycemic controls.170,184 Enhanced TNF-α levels have been directly linked to cellular changes in diabetes-enhanced periodontitis.196 TNF-α impairs the function of osteoblasts by blocking the differentiation of osteoblasts when inflammation is thought to be present. Furthermore, TNF-α can induce apoptosis by binding to TNF receptor-1, which triggers the initial events in apoptosis.158
It has been shown that increased bacterial infection-induced alveolar bone loss in diabetic subjects is accompanied by the enhanced expression of RAGE and inflammatory AGEs in gingival tissue.197 AGEs have been shown to interfere with osteoblast differentiation and to induce the apoptosis of osteoblasts in diabetes via the mitogen-activated protein kinase and cytosolic apoptotic pathway.89 Elevated levels of AGEs are found in the periodontium of diabetics, and the AGE–RAGE interaction results in an increase in pro-inflammatory cytokine expression and induces osteoblast apoptosis.186,198,199 CML-collagen, an AGE that is found in bone and serum, stimulates the apoptosis of bone-lining cells in vivo and in various osteoblastic cell cultures, which is mediated by RAGE.200
ROS production is another mechanism by which diabetes increases apoptosis. Persistent inflammation and hyperglycemia leads to cellular ROS accumulation, which is linked to diabetic complications.199,201 Moreover, increased oxidative stress in periodontal tissue has been shown to induce osteoblast apoptosis.202 It has been demonstrated that ROS causes the activation of caspase-3,203 which mediates osteoblast apoptosis.
Conclusion
In summary, diabetes mellitus and periodontal diseases are closely associated and exhibit similarities to other chronic diseases. Persistent hyperglycemia leading to exaggerated immune-inflammatory responses that are induced by periodontal pathogens is likely to be responsible for the greater risk and severity of periodontal disease in diabetics.134,199 Moreover, severe periodontitis that results in alveolar bone loss is likely to involve the effect of inflammation on both osteoclasts and osteoblasts. Diabetes has an important effect on enhancing osteoclastogenesis and on increasing osteoblast apoptosis. Interestingly, the effect of diabetes on bone loss and coupled bone formation is likely to involve the impact of diabetes on both the innate and adaptive immune responses.47,134
References
Bell GI, Polonsky KS . Diabetes mellitus and genetically programmed defects in beta-cell function . Nature 2001 ; 414 ( 6865 ): 788 – 791 .
Kahn SE . Clinical review 135: the importance of beta-cell failure in the development and progression of type 2 diabetes . J Clin Endocrinol Metab 2001 ; 86 ( 9 ): 4047 – 4058 .
Cavaghan MK, Ehrmann DA, Polonsky KS . Interactions between insulin resistance and insulin secretion in the development of glucose intolerance . J Clin Invest 2000 ; 106 ( 3 ): 329 – 333 .
Zimmet P, Alberti KG, Shaw J . Global and societal implications of the diabetes epidemic . Nature 2001 ; 414 ( 6865 ): 782 – 787 .
Kaul K, Tarr JM, Ahmad SI et al . Introduction to diabetes mellitus . Adv Exp Med Biol 2012 ; 771 : 1 – 11 .
American Diabetes Association . Diagnosis and classification of diabetes mellitus . Diabetes Care 2009 ; 32 ( Suppl 1 ): S62 – S67 .
SEARCH for Diabetes in Youth Study Group, Liese AD, D′Agostino RB Jr et al . The burden of diabetes mellitus among US youth: prevalence estimates from the SEARCH for Diabetes in Youth Study . Pediatrics 2006 ; 118 ( 4 ): 1510 – 1518 .
Soltesz G, Patterson CC, Dahlquist G et al . Worldwide childhood type 1 diabetes incidence—what can we learn from epidemiology? Pediatr Diabetes 2007 ; 8 ( Suppl 6 ): 6 – 14 .
Pinhas-Hamiel O, Zeitler P . The global spread of type 2 diabetes mellitus in children and adolescents . J Pediatr 2005 ; 146 ( 5 ): 693 – 700 .
Padgett LE, Broniowska KA, Hansen PA et al . The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis . Ann N Y Acad Sci 2013 ; 1281 : 16 – 35 .
Bluestone JA, Herold K, Eisenbarth G . Genetics, pathogenesis and clinical interventions in type 1 diabetes . Nature 2010 ; 464 ( 7293 ): 1293 – 1300 .
Anderson MS, Bluestone JA . The NOD mouse: a model of immune dysregulation . Annu Rev Immunol 2005 ; 23 : 447 – 485 .
van Belle TL, Coppieters KT, von Herrath MG . Type 1 diabetes: etiology, immunology, and therapeutic strategies . Physiol Rev 2011 ; 91 ( 1 ): 79 – 118 .
Nolan CJ, Damm P, Prentki M . Type 2 diabetes across generations: from pathophysiology to prevention and management . Lancet 2011 ; 378 ( 9786 ): 169 – 181 .
Prentki M, Nolan CJ . Islet beta cell failure in type 2 diabetes . J Clin Invest 2006 ; 116 ( 7 ): 1802 – 1812 .
Defronzo RA . Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus . Diabetes 2009 ; 58 ( 4 ): 773 – 795 .
Unger RH . Diabetic hyperglycemia: link to impaired glucose transport in pancreatic beta cells . Science 1991 ; 251 ( 4998 ): 1200 – 1205 .
Giacco F, Brownlee M . Oxidative stress and diabetic complications . Circ Res 2010 ; 107 ( 9 ): 1058 – 1070 .
Yerneni KK, Bai W, Khan BV et al . Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells . Diabetes 1999 ; 48 ( 4 ): 855 – 864 .
Kolm-Litty V, Sauer U, Nerlich A et al . High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells . J Clin Invest 1998 ; 101 ( 1 ): 160 – 169 .
Sayeski PP, Kudlow JE . Glucose metabolism to glucosamine is necessary for glucose stimulation of transforming growth factor-alpha gene transcription . J Biol Chem 1996 ; 271 ( 25 ): 15237 – 15243 .
Yan LJ . Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress . J Diabetes Res 2014 ; 2014 : 137919 .
Degenhardt TP, Thorpe SR, Baynes JW . Chemical modification of proteins by methylglyoxal . Cell Mol Biol: Noisy-le-grand 1998 ; 44 ( 7 ): 1139 – 1145 .
Wells-Knecht KJ, Zyzak DV, Litchfield JE et al . Mechanism of autoxidative glycosylation: identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose . Biochemistry 1995 ; 34 ( 11 ): 3702 – 3709 .
Thornalley PJ . The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life . Biochem J 1990 ; 269 ( 1 ): 1 – 11 .
Brownlee M . Biochemistry and molecular cell biology of diabetic complications . Nature 2001 ; 414 ( 6865 ): 813 – 820 .
Reaven GM, Hollenbeck C, Jeng CY et al . Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM . Diabetes 1988 ; 37 ( 8 ): 1020 – 1024 .
Walker KZ, O′Dea K, Johnson L et al . Body fat distribution and non-insulin-dependent diabetes: comparison of a fiber-rich, high-carbohydrate, low-fat (23%) diet and a 35% fat diet high in monounsaturated fat . Am J Clin Nutr 1996 ; 63 ( 2 ): 254 – 260 .
Maedler K, Oberholzer J, Bucher P et al . Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function . Diabetes 2003 ; 52 ( 3 ): 726 – 733 .
Maedler K, Spinas GA, Dyntar D et al . Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function . Diabetes 2001 ; 50 ( 1 ): 69 – 76 .
Unger RH . Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications . Diabetes 1995 ; 44 ( 8 ): 863 – 870 .
Cusi K . The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes . Curr Diab Rep 2010 ; 10 ( 4 ): 306 – 315 .
Evans JL, Goldfine ID, Maddux BA et al . Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes . Endocr Rev 2002 ; 23 ( 5 ): 599 – 622 .
Evans JL, Goldfine ID, Maddux BA et al . Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003 ; 52 ( 1 ): 1 – 8 .
Pitocco D, Zaccardi F, Di Stasio E et al . Oxidative stress, nitric oxide, and diabetes . Rev Diabet Stud 2010 ; 7 ( 1 ): 15 – 25 .
Niedowicz DM, Daleke DL . The role of oxidative stress in diabetic complications . Cell Biochem Biophys 2005 ; 43 ( 2 ): 289 – 330 .
Valle A, Giamporcaro GM, Scavini M et al . Reduction of circulating neutrophils precedes and accompanies type 1 diabetes . Diabetes 2013 ; 62 ( 6 ): 2072 – 2077 .
Harsunen MH, Puff R, D′Orlando O et al . Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes . Horm Metab Res 2013 ; 45 ( 6 ): 467 – 470 .
Battaglia M . Neutrophils and type 1 autoimmune diabetes . Curr Opin Hematol 2014 ; 21 ( 1 ): 8 – 15 .
Ding Y, Kantarci A, Hasturk H et al . Activation of RAGE induces elevated O2- generation by mononuclear phagocytes in diabetes . J Leukoc Biol 2007 ; 81 ( 2 ): 520 – 527 .
Bradshaw EM, Raddassi K, Elyaman W et al . Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells . J Immunol 2009 ; 183 ( 7 ): 4432 – 4439 .
Espinoza-Jiménez A, Peón AN, Terrazas LI . Alternatively activated macrophages in types 1 and 2 diabetes . Mediators Inflamm 2012 ; 2012 : 815953 .
Lontchi-Yimagou E, Sobngwi E, Matsha TE et al . Diabetes mellitus and inflammation . Curr Diab Rep 2013 ; 13 ( 3 ): 435 – 444 .
Cruz NG, Sousa LP, Sousa MO et al . The linkage between inflammation and Type 2 diabetes mellitus . Diabetes Res Clin Pract 2013 ; 99 ( 2 ): 85 – 92 .
Johnson DR, O′Connor JC, Satpathy A et al . Cytokines in type 2 diabetes . Vitam Horm 2006 ; 74 : 405 – 441 .
Hotamisligil GS, Shargill NS, Spiegelman BM . Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance . Science 1993 ; 259 ( 5091 ): 87 – 91 .
Nikolajczyk BS, Jagannathan-Bogdan M, Shin H et al . State of the union between metabolism and the immune system in type 2 diabetes . Genes Immun 2011 ; 12 ( 4 ): 239 – 250 .
Graves DT, Kayal RA . Diabetic complications and dysregulated innate immunity . Front Biosci 2008 ; 13 : 1227 – 1239 .
Pham MN, Kolb H, Battelino T et al . Fasting and meal-stimulated residual beta cell function is positively associated with serum concentrations of proinflammatory cytokines and negatively associated with anti-inflammatory and regulatory cytokines in patients with longer term type 1 diabetes . Diabetologia 2013 ; 56 ( 6 ): 1356 – 1363 .
Herder C, Carstensen M, Ouwens DM . Anti-inflammatory cytokines and risk of type 2 diabetes . Diabetes Obes Metab 2013 ; 15 ( Suppl 3 ): 39 – 50 .
Akash MS, Rehman K, Chen S . IL-1Ra and its delivery strategies: inserting the association in perspective . Pharm Res 2013 ; 30 ( 11 ): 2951 – 2966 .
Ardestani A, Sauter NS, Paroni F et al . Neutralizing interleukin-1beta (IL-1beta) induces beta-cell survival by maintaining PDX1 protein nuclear localization . J Biol Chem 2011 ; 286 ( 19 ): 17144 – 17155 .
Li S, Shin HJ, Ding EL et al . Adiponectin levels and risk of type 2 diabetes: a systematic review and meta-analysis . JAMA 2009 ; 302 ( 2 ): 179 – 188 .
Tandon N, Ali MK, Narayan KM . Pharmacologic prevention of microvascular and macrovascular complications in diabetes mellitus: implications of the results of recent clinical trials in type 2 diabetes . Am J Cardiovasc Drugs 2012 ; 12 ( 1 ): 7 – 22 .
Mattila TK, de Boer A . Influence of intensive versus conventional glucose control on microvascular and macrovascular complications in type 1 and 2 diabetes mellitus . Drugs 2010 ; 70 ( 17 ): 2229 – 2245 .
Gerstein HC, Pogue J, Mann JF et al . The relationship between dysglycaemia and cardiovascular and renal risk in diabetic and non-diabetic participants in the HOPE study: a prospective epidemiological analysis . Diabetologia 2005 ; 48 ( 9 ): 1749 – 1755 .
Laakso M . Hyperglycemia and cardiovascular disease in type 2 diabetes . Diabetes 1999 ; 48 ( 5 ): 937 – 942 .
Roy S, Sato T, Paryani G et al . Downregulation of fibronectin overexpression reduces basement membrane thickening and vascular lesions in retinas of galactose-fed rats . Diabetes 2003 ; 52 ( 5 ): 1229 – 1234 .
Chilelli NC, Burlina S, Lapolla A . AGEs, rather than hyperglycemia, are responsible for microvascular complications in diabetes: a “glycoxidation-centric” point of view . Nutr Metab Cardiovasc Dis 2013 ; 23 ( 10 ): 913 – 919 .
Behl Y, Krothapalli P, Desta T et al . Diabetes-enhanced tumor necrosis factor-alpha production promotes apoptosis and the loss of retinal microvascular cells in type 1 and type 2 models of diabetic retinopathy . Am J Pathol 2008 ; 172 ( 5 ): 1411 – 1418 .
Behl Y, Krothapalli P, Desta T et al . FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats . Diabetes 2009 ; 58 ( 4 ): 917 – 925 .
Davey GC, Patil SB, O′Loughlin A et al . Mesenchymal stem cell-based treatment for microvascular and secondary complications of diabetes mellitus . Front Endocrinol: Lausanne 2014 ; 5 : 86 .
Ferris FL 3rd, Davis MD, Aiello LM . Treatment of diabetic retinopathy . N Engl J Med 1999 ; 341 ( 9 ): 667 – 678 .
Han JW, Sin MY, Yoon YS . Cell therapy for diabetic neuropathy using adult stem or progenitor cells . Diabetes Metab J 2013 ; 37 ( 2 ): 91 – 105 .
Gooch C, Podwall D . The diabetic neuropathies . Neurologist 2004 ; 10 ( 6 ): 311 – 322 .
Duran-Salgado MB, Rubio-Guerra AF . Diabetic nephropathy and inflammation . World J Diabetes 2014 ; 5 ( 3 ): 393 – 398 .
Yamagishi S, Fukami K, Ueda S et al . Molecular mechanisms of diabetic nephropathy and its therapeutic intervention . Curr Drug Targets 2007 ; 8 ( 8 ): 952 – 959 .
Ziyadeh FN, Wolf G . Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy . Curr Diabetes Rev 2008 ; 4 ( 1 ): 39 – 45 .
Valkusz Z . [Diabetes and osteoporosis.] Orv Hetil 2011 ; 152 ( 29 ): 1161 – 1166 . Hungarian .
Vestergaard P, Rejnmark L, Mosekilde L . Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk . Diabetologia 2005 ; 48 ( 7 ): 1292 – 1299 .
Vestergaard P . Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—a meta-analysis . Osteoporos Int 2007 ; 18 ( 4 ): 427 – 444 .
Holmberg AH, Johnell O, Nilsson PM et al . Risk factors for fragility fracture in middle age. A prospective population-based study of 33 000 men and women . Osteoporos Int 2006 ; 17 ( 7 ): 1065 – 1077 .
Bonds DE, Larson JC, Schwartz AV et al . Risk of fracture in women with type 2 diabetes: the Women's Health Initiative Observational Study . J Clin Endocrinol Metab 2006 ; 91 ( 9 ): 3404 – 3410 .
Heap J, Murray MA, Miller SC et al . Alterations in bone characteristics associated with glycemic control in adolescents with type 1 diabetes mellitus . J Pediatr 2004 ; 144 ( 1 ): 56 – 62 .
Okazaki R . [Management of osteoporosis in diabetes mellitus.] Nippon Rinsho 2009 ; 67 ( 5 ): 1003 – 1010 . Japanese .
Schwartz AV, Hillier TA, Sellmeyer DE et al . Older women with diabetes have a higher risk of falls: a prospective study . Diabetes Care 2002 ; 25 ( 10 ): 1749 – 1754 .
Saito M, Fujii K, Mori Y et al . Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats . Osteoporos Int 2006 ; 17 ( 10 ): 1514 – 1523 .
Krakauer JC, McKenna MJ, Buderer NF et al . Bone loss and bone turnover in diabetes . Diabetes 1995 ; 44 ( 7 ): 775 – 782 .
Duarte VM, Ramos AM, Rezende LA et al . Osteopenia: a bone disorder associated with diabetes mellitus . J Bone Miner Metab 2005 ; 23 ( 1 ): 58 – 68 .
Williams GA, Callon KE, Watson M et al . Skeletal phenotype of the leptin receptor-deficient db/db mouse . J Bone Miner Res 2011 ; 26 ( 8 ): 1698 – 1709 .
Turner RT, Kalra SP, Wong CP et al . Peripheral leptin regulates bone formation . J Bone Miner Res 2013 ; 28 ( 1 ): 22 – 34 .
Suzuki K, Kurose T, Takizawa M et al . Osteoclastic function is accelerated in male patients with type 2 diabetes mellitus: the preventive role of osteoclastogenesis inhibitory factor/osteoprotegerin (OCIF/OPG) on the decrease of bone mineral density . Diabetes Res Clin Pract 2005 ; 68 ( 2 ): 117 – 125 .
Gerdhem P, Isaksson A, Akesson K et al . Increased bone density and decreased bone turnover, but no evident alteration of fracture susceptibility in elderly women with diabetes mellitus . Osteoporos Int 2005 ; 16 ( 12 ): 1506 – 1512 .
Norris R, Parker M . Diabetes mellitus and hip fracture: a study of 5966 cases . Injury 2011 ; 42 ( 11 ): 1313 – 1316 .
Perlman MH, Thordarson DB . Ankle fusion in a high risk population: an assessment of nonunion risk factors . Foot Ankle Int 1999 ; 20 ( 8 ): 491 – 496 .
Brown ML, Yukata K, Farnsworth CW et al . Delayed fracture healing and increased callus adiposity in a C57BL/6J murine model of obesity-associated type 2 diabetes mellitus . PLoS One 2014 ; 9 ( 6 ): e99656 .
Kayal RA, Tsatsas D, Bauer MA et al . Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity . J Bone Miner Res 2007 ; 22 ( 4 ): 560 – 568 .
Lozano D, de Castro LF, Dapía S et al . Role of parathyroid hormone-related protein in the decreased osteoblast function in diabetes-related osteopenia . Endocrinology 2009 ; 150 ( 5 ): 2027 – 2035 .
Alikhani M, Alikhani Z, Boyd C et al . Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways . Bone 2007 ; 40 ( 2 ): 345 – 353 .
Lu H, Kraut D, Gerstenfeld LC et al . Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation . Endocrinology 2003 ; 144 ( 1 ): 346 – 352 .
Hamann C, Goettsch C, Mettelsiefen J et al . Delayed bone regeneration and low bone mass in a rat model of insulin-resistant type 2 diabetes mellitus is due to impaired osteoblast function . Am J Physiol Endocrinol Metab 2011 ; 301 ( 6 ): E1220 – E1228 .
Weinberg E, Maymon T, Moses O et al . Streptozotocin-induced diabetes in rats diminishes the size of the osteoprogenitor pool in bone marrow . Diabetes Res Clin Pract 2014 ; 103 ( 1 ): 35 – 41 .
Santana RB, Xu L, Chase HB et al . A role for advanced glycation end products in diminished bone healing in type 1 diabetes . Diabetes 2003 ; 52 ( 6 ): 1502 – 1510 .
Schett G . Effects of inflammatory and anti-inflammatory cytokines on the bone . Eur J Clin Invest 2011 ; 41 ( 12 ): 1361 – 1366 .
Alblowi J, Kayal RA, Siqueira M et al . High levels of tumor necrosis factor-alpha contribute to accelerated loss of cartilage in diabetic fracture healing . Am J Pathol 2009 ; 175 ( 4 ): 1574 – 1585 .
Liu R, Bal HS, Desta T et al . Tumor necrosis factor-alpha mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing . Am J Pathol 2006 ; 168 ( 3 ): 757 – 764 .
Al-Mashat HA, Kandru S, Liu R et al . Diabetes enhances mRNA levels of proapoptotic genes and caspase activity, which contribute to impaired healing . Diabetes 2006 ; 55 ( 2 ): 487 – 495 .
Kayal RA, Siqueira M, Alblowi J et al . TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1 . J Bone Miner Res 2010 ; 25 ( 7 ): 1604 – 1615 .
Gilbert LC, Rubin J, Nanes MS . The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis . Am J Physiol Endocrinol Metab 2005 ; 288 ( 5 ): E1011 – E1018 .
Chang J, Liu F, Lee M et al . NF-κB inhibits osteogenic differentiation of mesenchymal stem cells by promoting β-catenin degradation . Proc Natl Acad Sci U S A 2013 ; 110 ( 23 ): 9469 – 9474 .
Lu X, Beck GR Jr, Gilbert LC et al . Identification of the homeobox protein Prx1 (MHox, Prrx-1) as a regulator of osterix expression and mediator of tumor necrosis factor α action in osteoblast differentiation . J Bone Miner Res 2011 ; 26 ( 1 ): 209 – 219 .
Stolzing A, Sellers D, Llewelyn O et al . Diabetes induced changes in rat mesenchymal stem cells . Cells Tissues Organs: Print 2010 ; 191 ( 6 ): 453 – 465 .
Kume S, Kato S, Yamagishi S et al . Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone . J Bone Miner Res 2005 ; 20 ( 9 ): 1647 – 1658 .
Notsu M, Yamaguchi T, Okazaki K et al . Advanced glycation end product 3 (AGE3) suppresses the mineralization of mouse stromal ST2 cells and human mesenchymal stem cells by increasing TGF-β expression and secretion . Endocrinology 2014 ; 155 ( 7 ): 2402 – 2410 .
Shin L, Peterson DA . Impaired therapeutic capacity of autologous stem cells in a model of type 2 diabetes . Stem Cells Transl Med 2012 ; 1 ( 2 ): 125 – 135 .
Takizawa M, Suzuki K, Matsubayashi T et al . Increased bone resorption may play a crucial role in the occurrence of osteopenia in patients with type 2 diabetes: possible involvement of accelerated polyol pathway in its pathogenesis . Diabetes Res Clin Pract 2008 ; 82 ( 1 ): 119 – 126 .
Reyes-García R, Rozas-Moreno P, López-Gallardo G et al . Serum levels of bone resorption markers are decreased in patients with type 2 diabetes . Acta Diabetol 2013 ; 50 ( 1 ): 47 – 52 .
Suzuki K, Ishida H, Takeshita N et al . Circulating levels of tartrate-resistant acid phosphatase in rat models of non-insulin-dependent diabetes mellitus . J Diabetes Complicat 1998 ; 12 ( 3 ): 176 – 180 .
Alblowi J, Tian C, Siqueira MF et al . Chemokine expression is upregulated in chondrocytes in diabetic fracture healing . Bone 2013 ; 53 ( 1 ): 294 – 300 .
Hie M, Shimono M, Fujii K et al . Increased cathepsin K and tartrate-resistant acid phosphatase expression in bone of streptozotocin-induced diabetic rats . Bone 2007 ; 41 ( 6 ): 1045 – 1050 .
Liu R, Bal HS, Desta T et al . Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation . J Dent Res 2006 ; 85 ( 6 ): 510 – 514 .
Jeffcoate WJ, Game F, Cavanagh PR . The role of proinflammatory cytokines in the cause of neuropathic osteoarthropathy (acute Charcot foot) in diabetes . Lancet 2005 ; 366 ( 9502 ): 2058 – 2061 .
Ha H, Kwak HB, Lee SW et al . Reactive oxygen species mediate RANK signaling in osteoclasts . Exp Cell Res 2004 ; 301 ( 2 ): 119 – 127 .
Drosatos-Tampakaki Z, Drosatos K, Siegelin Y et al . Palmitic acid and DGAT1 deficiency enhance osteoclastogenesis, while oleic acid-induced triglyceride formation prevents it . J Bone Miner Res 2014 ; 29 ( 5 ): 1183 – 1195 .
Catalfamo DL, Britten TM, Storch DL et al . Hyperglycemia induced and intrinsic alterations in type 2 diabetes-derived osteoclast function . Oral Dis 2013 ; 19 ( 3 ): 303 – 312 .
Miyata T, Kawai R, Taketomi S et al . Possible involvement of advanced glycation end-products in bone resorption . Nephrol Dial Transplant 1996 ; 11 ( Suppl 5 ): 54 – 57 .
Ding KH, Wang ZZ, Hamrick MW et al . Disordered osteoclast formation in RAGE-deficient mouse establishes an essential role for RAGE in diabetes related bone loss . Biochem Biophys Res Commun 2006 ; 340 ( 4 ): 1091 – 1097 .
Graves DT, Li J, Cochran DL . Inflammation and uncoupling as mechanisms of periodontal bone loss . J Dent Res 2011 ; 90 ( 2 ): 143 – 153 .
Borrell LN, Papapanou PN . Analytical epidemiology of periodontitis . J Clin Periodontol 2005 ; 32 ( Suppl 6 ): 132 – 158 .
Eke PI, Dye BA, Wei L et al . Prevalence of periodontitis in adults in the United States: 2009 and 2010 . J Dent Res 2012 ; 91 ( 10 ): 914 – 920 .
Dye BA . Global periodontal disease epidemiology . Periodontol 2000 2012 ; 58 ( 1 ): 10 – 25 .
Bascones-Martínez A, González-Febles J, Sanz-Esporrín J . Diabetes and periodontal disease. Review of the literature . Am J Dent 2014 ; 27 ( 2 ): 63 – 67 .
Pihlstrom BL, Michalowicz BS, Johnson NW . Periodontal diseases . Lancet 2005 ; 366 ( 9499 ): 1809 – 1820 .
Tanaka K, Miyake Y, Hanioka T et al . Relationship between IL1 gene polymorphisms and periodontal disease in Japanese women . DNA Cell Biol 2014 ; 33 ( 4 ): 227 – 233 .
Tsaousoglou P, Nietzsche S, Cachovan G et al . Antibacterial activity of moxifloxacin on bacteria associated with periodontitis within a biofilm . J Med Microbiol 2014 ; 63 ( Pt 2 ): 284 – 292 .
Jünemann S, Prior K, Szczepanowski R et al . Bacterial community shift in treated periodontitis patients revealed by ion torrent 16S rRNA gene amplicon sequencing . PLoS One 2012 ; 7 ( 8 ): e41606 .
Socransky SS, Haffajee AD, Cugini MA et al . Microbial complexes in subgingival plaque . J Clin Periodontol 1998 ; 25 ( 2 ): 134 – 144 .
Griffen AL, Beall CJ, Campbell JH et al . Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing . ISME J 2012 ; 6 ( 6 ): 1176 – 1185 .
Hajishengallis G, Liang S, Payne MA et al . Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement . Cell Host Microbe 2011 ; 10 ( 5 ): 497 – 506 .
Jiao Y, Hasegawa M, Inohara N . Emerging roles of immunostimulatory oral bacteria in periodontitis development . Trends Microbiol 2014 ; 22 ( 3 ): 157 – 163 .
Kumar PS, Griffen AL, Moeschberger ML et al . Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis . J Clin Microbiol 2005 ; 43 ( 8 ): 3944 – 3955 .
Kumar PS, Leys EJ, Bryk JM et al . Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing . J Clin Microbiol 2006 ; 44 ( 10 ): 3665 – 3673 .
Haffajee AD, Bogren A, Hasturk H et al . Subgingival microbiota of chronic periodontitis subjects from different geographic locations . J Clin Periodontol 2004 ; 31 ( 11 ): 996 – 1002 .
Graves DT, Oates T, Garlet GP . Review of osteoimmunology and the host response in endodontic and periodontal lesions . J Oral Microbiol 2011 ; 3 .
Boström L, Bergström J, Dahlén G et al . Smoking and subgingival microflora in periodontal disease . J Clin Periodontol 2001 ; 28 ( 3 ): 212 – 219 .
Williams RC, Jeffcoat MK, Kaplan ML et al . Flurbiprofen: a potent inhibitor of alveolar bone resorption in beagles . Science 1985 ; 227 ( 4687 ): 640 – 642 .
Assuma R, Oates T, Cochran D et al . IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis . J Immunol 1998 ; 160 ( 1 ): 403 – 409 .
Delima AJ, Karatzas S, Amar S et al . Inflammation and tissue loss caused by periodontal pathogens is reduced by interleukin-1 antagonists . J Infect Dis 2002 ; 186 ( 4 ): 511 – 516 .
Hajishengallis G . Aging and its impact on innate immunity and inflammation: implications for periodontitis . J Oral Biosci 2014 ; 56 ( 1 ): 30 – 37 .
Loos BG, Papantonopoulos G . Molecular biotypes for periodontal diseases? J Dent Res 2013 ; 92 ( 12 ): 1056 – 1057 .
Albert DA, Ward A, Allweiss P et al . Diabetes and oral disease: implications for health professionals . Ann N Y Acad Sci 2012 ; 1255 : 1 – 15 .
Bullon P, Newman HN, Battino M . Obesity, diabetes mellitus, atherosclerosis and chronic periodontitis: a shared pathology via oxidative stress and mitochondrial dysfunction? Periodontol 2000 2014 ; 64 ( 1 ): 139 – 153 .
Tsai C, Hayes C, Taylor GW . Glycemic control of type 2 diabetes and severe periodontal disease in the US adult population . Community Dent Oral Epidemiol 2002 ; 30 ( 3 ): 182 – 192 .
Preshaw PM, Bissett SM . Periodontitis: oral complication of diabetes . Endocrinol Metab Clin North Am 2013 ; 42 ( 4 ): 849 – 867 .
Löe H . Periodontal disease. The sixth complication of diabetes mellitus . Diabetes Care 1993 ; 16 ( 1 ): 329 – 334 .
Popławska-Kita A, Siewko K, Szpak P et al . Association between type 1 diabetes and periodontal health . Adv Med Sci 2014 ; 59 ( 1 ): 126 – 131 .
Xavier AC, Silva IN, Costa Fde O et al . [Periodontal status in children and adolescents with type 1 diabetes mellitus.] Arq Bras Endocrinol Metabol 2009 ; 53 ( 3 ): 348 – 354 . Portuguese .
Leite RS, Marlow NM, Fernandes JK . Oral health and type 2 diabetes . Am J Med Sci 2013 ; 345 ( 4 ): 271 – 273 .
Fernandes JK, Wiegand RE, Salinas CF et al . Periodontal disease status in gullah african americans with type 2 diabetes living in South Carolina . J Periodontol 2009 ; 80 ( 7 ): 1062 – 1068 .
Al-Khabbaz AK . Type 2 diabetes mellitus and periodontal disease severity . Oral Health Prev Dent 2014 ; 12 ( 1 ): 77 – 82 .
Apoorva SM, Sridhar N, Suchetha A . Prevalence and severity of periodontal disease in type 2 diabetes mellitus (non-insulin-dependent diabetes mellitus) patients in Bangalore city: an epidemiological study . J Indian Soc Periodontol 2013 ; 17 ( 1 ): 25 – 29 .
Ryan ME, Carnu O, Kamer A . The influence of diabetes on the periodontal tissues . J Am Dent Assoc 2003 ; 134 (Spec No) : 34S – 40S .
Orbak R, Simsek S, Orbak Z et al . The influence of type-1 diabetes mellitus on dentition and oral health in children and adolescents . Yonsei Med J 2008 ; 49 ( 3 ): 357 – 365 .
Costa CC, Resende GB, Souza JM et al . [Study of the oral manifestations in diabetic children and their correlation variables.] Arq Bras Endocrinol Metabol 2004 ; 48 ( 3 ): 374 – 378 . Portuguese .
Albandar JM, Tinoco EM . Global epidemiology of periodontal diseases in children and young persons . Periodontol 2000 2002 ; 29 : 153 – 176 .
Katagiri S, Nagasawa T, Kobayashi H et al . Improvement of glycemic control after periodontal treatment by resolving gingival inflammation in type 2 diabetic patients with periodontal disease . J Diabetes Investig 2012 ; 3 ( 4 ): 402 – 409 .
Kang J, de Brito Bezerra B, Pacios S et al . Aggregatibacter actinomycetemcomitans infection enhances apoptosis in vivo through a caspase-3-dependent mechanism in experimental periodontitis . Infect Immun 2012 ; 80 ( 6 ): 2247 – 2256 .
Graves DT, Liu R, Oates TW . Diabetes-enhanced inflammation and apoptosis: impact on periodontal pathosis . Periodontol 2000 2007 ; 45 : 128 – 137 .
Ponugoti B, Dong G, Graves DT . Role of forkhead transcription factors in diabetes-induced oxidative stress . Exp Diabetes Res 2012 ; 2012 : 939751 .
Andriankaja OM, Galicia J, Dong G et al . Gene expression dynamics during diabetic periodontitis . J Dent Res 2012 ; 91 ( 12 ): 1160 – 1165 .
Pacios S, Andriankaja O, Kang J et al . Bacterial infection increases periodontal bone loss in diabetic rats through enhanced apoptosis . Am J Pathol 2013 ; 183 ( 6 ): 1928 – 1935 .
Mealey BL, Rose LF . Diabetes mellitus and inflammatory periodontal diseases . Curr Opin Endocrinol Diabetes Obes 2008 ; 15 ( 2 ): 135 – 141 .
Lalla E, Cheng B, Lal S et al . Diabetes mellitus promotes periodontal destruction in children . J Clin Periodontol 2007 ; 34 ( 4 ): 294 – 298 .
Deshpande K, Jain A, Sharma R et al . Diabetes and periodontitis . J Indian Soc Periodontol 2010 ; 14 ( 4 ): 207 – 212 .
Tervonen T, Karjalainen K, Knuuttila M et al . Alveolar bone loss in type 1 diabetic subjects . J Clin Periodontol 2000 ; 27 ( 8 ): 567 – 571 .
Kim JH, Lee DE, Gunawardhana KS et al . Effect of the interaction between periodontitis and type 1 diabetes mellitus on alveolar bone, mandibular condyle and tibia . Acta Odontol Scand 2014 ; 72 ( 4 ): 265 – 273 .
Kim JH, Lee DE, Choi SH et al . Diabetic characteristics and alveolar bone loss in streptozotocin- and streptozotocin-nicotinamide-treated rats with periodontitis . J Periodont Res 2014 ; 49 ( 6 ): 792 – 800 .
Taylor GW, Burt BA, Becker MP et al . Non-insulin dependent diabetes mellitus and alveolar bone loss progression over 2 years . J Periodontol 1998 ; 69 ( 1 ): 76 – 83 .
Taylor GW, Burt BA, Becker MP et al . Glycemic control and alveolar bone loss progression in type 2 diabetes . Ann Periodontol 1998 ; 3 ( 1 ): 30 – 39 .
Pacios S, Kang J, Galicia J et al . Diabetes aggravates periodontitis by limiting repair through enhanced inflammation . FASEB J 2012 ; 26 ( 4 ): 1423 – 1430 .
Silva JA, Lopes Ferrucci D, Peroni LA et al . Periodontal disease-associated compensatory expression of osteoprotegerin is lost in type 1 diabetes mellitus and correlates with alveolar bone destruction by regulating osteoclastogenesis . Cells Tissues Organs: Print 2012 ; 196 ( 2 ): 137 – 150 .
Graves DT, Naguib G, Lu H et al . Inflammation is more persistent in type 1 diabetic mice . J Dent Res 2005 ; 84 ( 4 ): 324 – 328 .
Naguib G, Al-Mashat H, Desta T et al . Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation . J Invest Dermatol 2004 ; 123 ( 1 ): 87 – 92 .
Hasturk H, Kantarci A, Goguet-Surmenian E et al . Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo . J Immunol 2007 ; 179 ( 10 ): 7021 – 7029 .
Andriankaja OM, Galicia J, Dong G et al . Gene expression dynamics during diabetic periodontitis . J Dent Res 2012 ; 91 ( 12 ): 1160 – 1165 .
Southerland JH, Taylor GT, Offenbacher S . Diabetes and periodontal infection: making the connection . Clin Diab 2005 ; 23 ( 4 ): 171 – 178 .
Silva JA, Ferrucci DL, Peroni LA et al . Sequential IL-23 and IL-17 and increased Mmp8 and Mmp14 expression characterize the progression of an experimental model of periodontal disease in type 1 diabetes . J Cell Physiol 2012 ; 227 ( 6 ): 2441 – 2450 .
Duarte PM, de Oliveira MC, Tambeli CH et al . Overexpression of interleukin-1beta and interleukin-6 may play an important role in periodontal breakdown in type 2 diabetic patients . J Periodont Res 2007 ; 42 ( 4 ): 377 – 381 .
Bastos AS, Graves DT, Loureiro AP et al . Lipid peroxidation is associated with the severity of periodontal disease and local inflammatory markers in patients with type 2 diabetes . J Clin Endocrinol Metab 2012 ; 97 ( 8 ): E1353 – E1362 .
Lappin DF, Eapen B, Robertson D et al . Markers of bone destruction and formation and periodontitis in type 1 diabetes mellitus . J Clin Periodontol 2009 ; 36 ( 8 ): 634 – 641 .
Mahamed DA, Marleau A, Alnaeeli M et al . G− anaerobes-reactive CD4+ T-cells trigger RANKL-mediated enhanced alveolar bone loss in diabetic NOD mice . Diabetes 2005 ; 54 ( 5 ): 1477 – 1486 .
Santos VR, Lima JA, Gonçalves TE et al . Receptor activator of nuclear factor-kappa B ligand/osteoprotegerin ratio in sites of chronic periodontitis of subjects with poorly and well-controlled type 2 diabetes . J Periodontol 2010 ; 81 ( 10 ): 1455 – 1465 .
Brownlee M, Cerami A, Vlassara H . Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications . N Engl J Med 1988 ; 318 ( 20 ): 1315 – 1321 .
Zizzi A, Tirabassi G, Aspriello SD et al . Gingival advanced glycation end-products in diabetes mellitus-associated chronic periodontitis: an immunohistochemical study . J Periodont Res 2013 ; 48 ( 3 ): 293 – 301 .
Lamster IB . Diabetes mellitus and oral health: an interprofessional approach . Ames : John Wiley & Sons, 2014 : 130 .
Mealey BL, Oates TW, American Academy of Periodontology . Diabetes mellitus and periodontal diseases . J Periodontol 2006 ; 77 ( 8 ): 1289 – 1303 .
Sies H . Oxidative stress: oxidants and antioxidants . Exp Physiol 1997 ; 82 ( 2 ): 291 – 295 .
Steinbeck MJ, Appel WH Jr, Verhoeven AJ et al . NADPH-oxidase expression and in situ production of superoxide by osteoclasts actively resorbing bone . J Cell Biol 1994 ; 126 ( 3 ): 765 – 772 .
Thomas B, Ramesh A, Suresh S et al . A comparative evaluation of antioxidant enzymes and selenium in the serum of periodontitis patients with diabetes mellitus type 2 . Contemp Clin Dent 2013 ; 4 ( 2 ): 176 – 180 .
Garrett IR, Boyce BF, Oreffo RO et al . Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo . J Clin Invest 1990 ; 85 ( 3 ): 632 – 639 .
Collin HL, Sorsa T, Meurman JH et al . Salivary matrix metalloproteinase (MMP-8) levels and gelatinase (MMP-9) activities in patients with type 2 diabetes mellitus . J Periodont Res 2000 ; 35 ( 5 ): 259 – 265 .
Fu YW, He HB, Ou JG . [Osteoblast apoptosis in experimental diabetic periodontitis in rats.] Hua Xi Kou Qiang Yi Xue Za Zhi 2009 ; 27 ( 3 ): 252 – 255, 259 . Chinese .
Behl Y, Siqueira M, Ortiz J et al . Activation of the acquired immune response reduces coupled bone formation in response to a periodontal pathogen . J Immunol 2008 ; 181 ( 12 ): 8711 – 8718 .
Isaka J, Ohazama A, Kobayashi M et al . Participation of periodontal ligament cells with regeneration of alveolar bone . J Periodontol 2001 ; 72 ( 3 ): 314 – 323 .
He H, Liu R, Desta T et al . Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss . Endocrinology 2004 ; 145 ( 1 ): 447 – 452 .
Naguib G, Al-Mashat H, Desta T et al . Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation . J Invest Dermatol 2004 ; 123 ( 1 ): 87 – 92 .
Lalla E, Lamster IB, Schmidt AM . Enhanced interaction of advanced glycation end products with their cellular receptor RAGE: implications for the pathogenesis of accelerated periodontal disease in diabetes . Ann Periodontol 1998 ; 3 ( 1 ): 13 – 19 .
Taylor JJ, Preshaw PM, Lalla E . A review of the evidence for pathogenic mechanisms that may link periodontitis and diabetes . J Periodontol 2013 ; 84 ( 4 Suppl ): S113 – S134 .
Chapple IL, Genco R, working group 2 of the joint EFP/AAP workshop . Diabetes and periodontal diseases: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases . J Periodontol 2013 ; 84 ( 4 Suppl ): S106 – S112 .
Hein G, Weiss C, Lehmann G et al . Advanced glycation end product modification of bone proteins and bone remodelling: hypothesis and preliminary immunohistochemical findings . Ann Rheum Dis 2006 ; 65 ( 1 ): 101 – 104 .
Fu YW, He HB . Apoptosis of periodontium cells in streptozototocin- and ligature-induced experimental diabetic periodontitis in rats . Acta Odontol Scand 2013 ; 71 ( 5 ): 1206 – 1215 .
Ohnishi T, Bandow K, Kakimoto K et al . Oxidative stress causes alveolar bone loss in metabolic syndrome model mice with type 2 diabetes . J Periodont Res 2009 ; 44 ( 1 ): 43 – 51 .
Wang GW, Klein JB, Kang YJ . Metallothionein inhibits doxorubicin-induced mitochondrial cytochrome c release and caspase-3 activation in cardiomyocytes . J Pharmacol Exp Ther 2001 ; 298 ( 2 ): 461 – 468 .
Acknowledgements
The authors would like to thank Sunitha Batchu for help in preparing this manuscript and Megha Dalalfor for technical assistance. This work was funded by the National Institute of Dental and Craniofacial Research (grant no. DE021921).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Wu, YY., Xiao, E. & Graves, D. Diabetes mellitus related bone metabolism and periodontal disease. Int J Oral Sci 7, 63–72 (2015). https://doi.org/10.1038/ijos.2015.2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijos.2015.2
Keywords
This article is cited by
-
Severe periodontal disease in Japanese patients with high HbA1c levels: a cross-sectional study
Diabetology International (2024)
-
Dyslipidemia, diabetes and periodontal disease, a cross-sectional study in Rafsanjan, a region in southeast Iran
BMC Oral Health (2023)
-
Association of oral health with various work problems: a cross-sectional study of Japanese workers
BMC Oral Health (2023)
-
Analysis of multiple chronic disease characteristics in middle-aged and elderly South Koreans by exercise habits based on association rules mining algorithm
BMC Public Health (2023)
-
A novel modified-curcumin 2.24 resolves inflammation by promoting M2 macrophage polarization
Scientific Reports (2023)
