
Abstract
Purpose:
We aimed to identify the genetic cause in a cohort of 11 unrelated cases and two sisters with 46,XX SRY-negative (ovo)testicular disorders of sex development (DSD).
Methods:
Whole-exome sequencing (n = 9), targeted resequencing (n = 4), and haplotyping were performed. Immunohistochemistry of sex-specific markers was performed on patients’ gonads. The consequences of mutation were investigated using luciferase assays, localization studies, and RNA-seq.
Results:
We identified a novel heterozygous NR5A1 mutation, c.274C>T p.(Arg92Trp), in three unrelated patients. The Arg92 residue is highly conserved and located in the Ftz-F1 region, probably involved in DNA-binding specificity and stability. There were no consistent changes in transcriptional activation or subcellular localization. Transcriptomics in patient-derived lymphocytes showed upregulation of MAMLD1, a direct NR5A1 target previously associated with 46,XY DSD. In gonads of affected individuals, ovarian FOXL2 and testicular SRY-independent SOX9 expression observed.
Conclusions:
We propose NR5A1, previously associated with 46,XY DSD and 46,XX primary ovarian insufficiency, as a novel gene for 46,XX (ovo)testicular DSD. We hypothesize that p.(Arg92Trp) results in decreased inhibition of the male developmental pathway through downregulation of female antitestis genes, thereby tipping the balance toward testicular differentiation in 46,XX individuals. In conclusion, our study supports a role for NR5A1 in testis differentiation in the XX gonad.
Genet Med 19 4, 367–376.
Similar content being viewed by others


Introduction
Although several genetic mechanisms leading to 46,XY disorders of sex development (DSD) have been elucidated in recent years, little is known about the developmental pathways that may induce testicular development in 46,XX individuals when dysregulated. Approximately 80% of all 46,XX testicular DSD cases are explained by a translocation of the Sex Determining Region Y (SRY) gene.1,2 Duplications of SRY-box 9 (SOX9) or its regulatory region are a rare cause of 46,XX testicular DSD.3,4 Copy number variations disrupting the regulatory region of SRY-box 3 (SOX3), a putative ancestor of SRY, have also been found in patients with 46,XX DSD.5,6 In line with this, 46,XX DSD phenotypes have been associated with partial duplications of human chromosome 22q13 containing SRY-box 10 (SOX10).7,8 Studies in mice overexpressing Sox10 strongly implicate gain-of-function of SOX10 in 46,XX DSD.9 In addition to these gain-of-function changes of male sex-determining genes, loss-of-function mutations of female sex-determining genes can give rise to 46,XX DSD. This is illustrated by biallelic loss-of-function mutations in R-Spondin 1 (RSPO1), which have been reported in a rare syndrome characterized by 46,XX testicular DSD, palmoplantar hyperkeratosis, and susceptibility to development of squamous cell carcinoma.10,11
Interestingly, intrafamilial phenotypical variability and incomplete penetrance have been described, ranging from unaffected mutation carriers to cases with 46,XX testicular DSD or ovotesticular DSD, indicating that these conditions may represent a phenotypic spectrum of the same underlying defect.12,13,14
Here, we aimed to identify the underlying molecular genetic cause in 11 unrelated individuals and 2 sisters with 46,XX testicular and ovotesticular DSD; the latter was defined as the combined presence of testis and ovarian tissue in the same individual. In three of them, we identified a novel and recurrent heterozygous variant, c.274C>T p.(Arg92Trp), in the Nuclear Receptor family 5 subfamily A member 1 gene (NR5A1, AD4BP). The impact of this variant was further studied by structural remodeling of the affected amino acid, luciferase and localization studies in cellular systems, RNA-seq on patient-derived lymphocytes, and immunohistochemistry of patients’ gonads.
Materials and Methods
Subjects
Case 1, the second child of two healthy, nonconsanguineous parents, was born after an uneventful pregnancy. Mild hypertrophy of the clitoris was noticed at birth. Vaginoscopy and laparoscopy detected the presence of a cervix and possibly a right hemiuterus, a left epididymis, and male vasculature. Both inguinal gonads were biopsied. The left gonad revealed testicular differentiation with germ cells present; the right gonad contained only fibrotic tissue. A human chorionic gonadotropin (hCG) stimulation test (1,500 IU administrated intramuscularly with blood sampling after 72 h) was performed on the seventh day after birth, resulting in a testosterone level of 2.52 nmol/l (baseline: 0.72 nmol/l); anti-Müllerian hormone (AMH) was between the male and female references (99.9 pmol/l). Based on these results and the child’s phenotype, a female sex was assigned. Microarray-based comparative genomic hybridization (array CGH) showed a normal female pattern, and fluorescence in situ hybridization (FISH) with SRY-specific probes excluded SRY translocation. The results of quantitative polymerase chain reaction for the regulatory RevSex locus, upstream of SOX9, and MLPA for different sex development–related genes (P185 Intersex, MRC Holland, Amsterdam, The Netherlands) were normal.
Case 2 is a 22-year-old woman who had been diagnosed with 46,XX ovotesticular DSD. She had ambiguous genitalia at birth and a short, blind-ending vagina. At age 4, she underwent clitoroplasty and bilateral gonadectomy of abdominal gonads, which both turned out to be ovotestes, including primordial and primary follicles in the ovarian part and Sertoli cell–only tubules in the testicular part. SRY-specific FISH was negative, and array CGH revealed a small duplication reported to be a benign variant (hg19: chr 8: 132161619 – 132812388). RevSex locus-specific quantitative polymerase chain reaction was normal. She has one younger sister, who displayed normal puberty; family history was unremarkable.
Case 3 is a 23-year-old man with 46,XX testicular DSD. At birth, a micropenis, penile hypospadias, and bilateral scrotal but atrophic testes were noticed. Gonadal biopsies at the age of 2 years revealed bilateral testicular differentiation with immature seminiferous tubules without germ cells and limited intertubular fibrosis. When he was 12 years old, a prolonged hCG test (2 × 1,500 IU per week for 3 consecutive weeks) led to a peak increase of serum testosterone to 6.0 nmol/l. Hypergonadotrophic hypogonadism developed rapidly thereafter, and testosterone supplementation was started at the age of 13. FISH with SRY and TSPY probes was negative, and array CGH and quantitative polymerase chain reaction for the RevSex locus were normal. He is the oldest of three brothers; familial history is unremarkable.
All three patients are Caucasian. Cases 1 and 3 are from East Flanders in Belgium, and case 2 originates from the south of the Netherlands, close to the Belgian-Dutch border. All families live near (within 70 km) each other. Pedigrees of the three families are shown in Figure 1a and phenotypic data for cases 1–3 are summarized in Table 1 ; phenotypic data for cases 4–9 are presented in the Supplementary Data online (Supplementary Table S1 online).

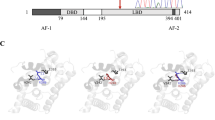
Pedigrees and haplotypes of mutation-positive families and schematic representation of the NR5A1 protein, mutation location, and conservation. (a) Pedigrees for families 1–3. Affected individuals heterozygous for NR5A1 mutation c.274C>T p.(Arg92Trp) are indicated by filled symbols. Asymptomatic individuals with NR5A1 mutation p.(Arg92Trp) are represented by a black spot in the symbol. In total, 29 microsatellite markers were used to assess the haplotypes; only the ones building up the common haplotype and flanking markers are shown here. The common haplotype (in red box) shared by the three mutation carriers has a maximum size of 1.5 Mb. (b) The different domains found in NR5A1, a DNA-binding domain at the C-terminus, followed by the Ftz-F1 region, which is important for DNA-binding stability, a hinge region, and a ligand-binding domain. An electropherogram showing the c.274C>T substitution is depicted below. The protein alignment of p.(Arg92Trp) and its surrounding amino acids is shown on top. The affected amino acid and the surrounding region are conserved up to zebrafish.
Genetic study
Whole-exome sequencing (WES) was performed in nine patients with 46,XX (ovo)testicular DSD; in four additional patients, included later, the coding region of NR5A1 was sequenced. Enrichment for WES was performed with the SureSelectXT Human All Exon V5 kit (Agilent), followed by paired-end sequencing on HiSeq 2000 (2 × 100 cycles) (Illumina). Reads were mapped against the human reference genome sequence (NCBI, GRCh37/hg19) with the CLC Genomics Workbench v6.4 (Qiagen), followed by postmapping duplicate-read removal, coverage analysis, and quality-based variant calling. More thorough variant annotation was executed with Alamut-HT v1.1.5 and Alamut Visual v2.7 software (Interactive Biosoftware). For case 1, because we had access to parental DNA, a trio-sequencing strategy was used. Variant filtering was performed with the Ingenuity Variant Analysis platform (Qiagen). Sanger sequencing was used to confirm potentially pathogenic variants identified by WES and to perform segregation analysis. Primers were designed with primer3plus. Sequencing was performed on the ABI 3730XL DNA Analyzer (Applied Biosystems) using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), followed by data analysis with SeqScape v2.5 (Life Technologies). Microsatellite analysis with 29 markers (deCODE, Généthon, and Marshfield) was performed to assess disease haplotypes in the three mutation-positive cases. Data analysis was performed with the GeneMapper v3.7 software (Applied Biosystems).
In silico modeling of the p.(Arg92Trp) and p.(Arg92Gln) mutations
All modeling and calculations were performed using model 1 of the NMR structure of the NR5A1 DNA binding domain (PDB code: 2ff0).15 The effect of the mutations on the stability of the protein was calculated using the FoldX RepairPDB and BuildModel commands, with 20 replicate calculations.16 Models for p.(Arg92Trp) and, for comparison, p.(Arg92Gln) mutants of the NR5A1 DNA-binding domain in complex with DNA were built using the YASARA structure with the swap and optimize commands to replace R92 by a tryptophan or glutamine, followed by energy minimization with the YASARA forcefield in explicit solvent.17 The in silico binding energy between the NR5A1 LBD and the DNA fragments was calculated using the YASARA BindEnergy command.17 Models and structures were visualized via UCSF chimera.18
Plasmid construction
We constructed a GFP-tagged NR5A1 wild-type (WT) construct starting from the Gateway pcDNA-DEST47 vector (Invitrogen, Life Technologies) and a WT NR5A1 open reading frame (ORF)-containing entry clone using a Gateway LR-recombination reaction (Invitrogen, Life Technologies). The mutations undergoing study were inserted using the Q5 site-directed mutagenesis kit (New England Biolabs), and mutation-specific primers were designed with the NEBaseChanger software (New England Biolabs). Mutated plasmids were transformed in One Shot TOP10 competent cells (Invitrogen, Life Technologies) and subsequently isolated with the NucleoBond Xtra Midi/Maxi kit (Macherey-Nagel). The entire NR5A1 insert and the surrounding backbone were sequenced and selected plasmids were grown to obtain larger quantities.
Luciferase assays
Luciferase assays were performed in two cell lines: HeLa and KGN. Cells were grown in DMEM-F12 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. One day before transfection, 5,000 cells/well were seeded in a 96-well plate. Each experiment was performed in six independent replicates. Cells were transfected with 187 ng of a DNA mixture using calcium phosphate transfection, after which they were washed with phosphate-buffered saline and ethylene glycol tetra-acetic acid to remove remaining precipitates. After 48 h, cells were washed with phosphate-buffered saline again before lysis and luciferase activities were assessed with the Dual-Glo Luciferase Assay System (Promega). Results are reported as relative light units (R.L.U., i.e., the ratio of the Renilla luciferase to firefly luciferase activities). Each value is the mean of six independent experiments, and the standard error bar represents the standard error of the mean.
Subcellular localization
Cells were seeded in 24-well plates containing sterile coverslips at a concentration of 30,000 cells per well 1 day before transfection. A total of 1,250 ng DNA per well was transfected using the calcium phosphate method. At 48 h after transfection, cells were washed with phosphate-buffered saline and fixed with 4% paraformaldehyde at room temperature for 15 min. Nuclei were stained with Hoechst (1/500) and coverslips were mounted on slides with Vectashield mounting medium (Vector Laboratories). Subcellular localization was assessed using fluorescence microscopy. All transfections were performed in duplicate, and at least 100 cells were counted per transfection.
RNA-seq
RNA-seq was performed on cultured lymphocytes from two patients (cases 1 and 3) and four female control samples. Lymphocytes were isolated from EDTA blood using Lymphoprep (Stemcell Technologies), cultured in Roswell Park Memorial Institute medium with 10% fetal bovine serum, and substituted with interleukin-2 and phytohemagglutinin. RNA was isolated with the QiaAmp RNeasy Mini-Kit (Qiagen), followed by sample preparation with the TruSeq Stranded mRNA sample preparation kit (Illumina), subsequent cluster generation, and single-end sequencing with the NextSeq 500 High Output Kit V2 (75 cycles) (Illumina). To obtain equally large patient and control groups, patient samples were run in duplicate. First, all reads were mapped against the human GRCh38 reference genome with TopHat (v.2.1.0),19 followed by transcriptome assembly with Cufflinks (v.2.2.1).20 Subsequently, a read count matrix was generated using HTSeq-count (v.0.6.1p1).21 Trimmed mean of M-values normalization and differential gene expression were assessed with the Limma software package.22 The statistical significance of each observed change in expression was tested between the patient group (two patient samples, two replicates per patient) and the control group (four control samples) with a false discovery rate threshold of 5%.
Immunohistochemistry
Slides with thicknesses of 4 µm were prepared from formalin-fixed (10%), paraffin-embedded tissue and pretreated for 5 min with 3% H2O2 to block endogenous peroxidase. After heat-induced antigen retrieval at pH 9, slides were blocked for biotin (SP-2001, Vector Laboratories, Burlingame, CA). After overnight incubation at 4 °C with SOX9 (dilution 1/250, AF3075, R&D systems), FOXL2 (dilution 1/350, a gift from Marc Fellous), and DDX4 primary antibody (dilution 1/100, Ab13840, Abcam), detection was performed by 30-min incubation at room temperature with a biotin-labeled secondary antibody, followed by another 30-min incubation with horseradish peroxidase (HRP)-conjugated Avidin-Biotin complex. Visualization was performed using 3,3′-diaminobenzidine (DAB)/H202, (SOX9 and DDX4), and aminoethyl carbazole (AEC)/H202 (FOXL2); counterstaining was performed with hematoxylin. General morphology was assessed on adjacent slides after hematoxylin and eosin staining.
Results
Novel and recurrent missense mutation in NR5A1 in three patients with 46,XX (Ovo)Testicular DSD
Eleven unrelated patients and two sisters with 46,XX ovotesticular or testicular DSD were included in this study. Nine underwent WES and four underwent resequencing of the coding region of NR5A1 because they were included at a later time. When exome data were available, all variants were filtered against a list of known 46,XX and 46,XY DSD genes (Supplementary Data online, Supplementary Table S2 online), annotated, and manually curated. All variants in DSD genes, identified in cases 1–9, are listed in Supplementary Data online and Supplementary Tables S3–S11 online. We identified a heterozygous missense variant in exon 4 of NR5A1 c.274C>T p.(Arg92Trp) in cases 1–3. Prediction programs SIFT, Polyphen-2, and MutationTaster suggest a possible effect on protein function. The physicochemical distance between Arg and Trp is moderate (Grantham score of 101) and the variant is absent in genomic databases such as dbSNP, ESP, ExAC, GoNL, and 1000 Genomes and in an in-house exome database. The affected amino acid and surrounding region are highly conserved up to zebrafish and is part of the Ftz-F1 region ( Figure 1b ).23 Segregation analysis of the three families showed incomplete penetrance in four 46,XX family members ( Figure 1a ). In case 1, the mutation was paternally inherited (family 1: II:2); in the other two families, the mutation was transmitted by the mother (family 2: I:2; family 3: I:2). The gynecological history for these unaffected women as well as for the older sister of case 1, who also carries the mutation (family 1: III.1), was unremarkable (Supplementary Data online, Supplementary Table S12 online). Microsatellite analysis showed a haplotype of approximately 1.5 Mb shared among cases 1–3 ( Figure 1a ), pointing to a founder effect. Given the incomplete penetrance, we assessed the WES data for other variants that might contribute to the phenotype. Variant lists with de novo variants for case 1 and rare variants (minor allele frequency < 0.01) for cases 2 and 3 were added to the Supplementary Data online and Supplementary Tables S13–S16 online.
Protein structure modeling of NR5A1 p.(Arg92Trp) and p.(Arg92Gln)
We performed protein structural modeling, starting from a model of NR5A1 bound to the α-inhibin promoter, for the following mutations: p.(Arg92Trp) found here in the three cases with 46,XX DSD and p.(Arg92Gln) previously found in 46,XY DSD.24 FoldX calculations predict that the mutations do not affect the stability of the isolated NR5A1 DNA-binding domain.16 In the models of mutants bound to DNA, the tryptophan and glutamine side chains do not penetrate as deeply into the minor groove as does the wild-type arginine side chain. Although the Arg92 side chain makes a hydrogen bond with a deoxycytosine base, no hydrogen bonds are formed by the mutant side chains. The (enthalpic) in silico binding energy for interaction of the DNA-binding domain of NR5A1 with the DNA segment is 9% lower for both mutants. This suggests that both mutations can affect the interaction of NR5A1 with its response elements, although effects may be variable and specific for different response elements ( Figure 2 ).

Structural analysis of NR5A1 (PDB code 2FF0). Position of Arg92 (R92) in the wild-type NR5A1 DNA-binding domain (DBD). (a) R92 is part of the C-terminal A-box and its side chain fits in the minor groove, making a hydrogen bond with a deoxycytosine base. Models of the p.(Arg92Trp) (Trp=W) (b) and p.(Arg92Gln) (Gln=Q) (c) mutants show how this mutated side chain binds in the DNA minor groove without hydrogen bond formation to the DNA. The affected amino acid R92 is located in a region C-terminal to the classical DBD with its two zinc (Zn) fingers. This additional domain is found in family members of NR5A1, including NGFI-B, and was called the A-box or Ftz-F1 box.15 This A-box domain has been implicated in interactions with the minor groove of the target DNA. Prior to structure determination, binding studies with NR5A1 and NGFI-B mutants showed that this interaction is important for their binding specificity toward different DNA targets. For NR5A1, this specificity is determined by amino acids 91–93, which are predicted to interact with three specific basepairs 5ʹ of the classical half-site bound by the Zn fingers. The results of this study are in line with those for the later NMR structure of the NR5A1 DBD bound to a DNA fragment of the inhibin-alpha subunit promoter. In this structure, the side chain of R92 inserts deeply into the minor groove, and its guanidine head group binds to two of the three basepairs at the 5ʹ of classical half-sites. Mutation R92A abolished DNA-binding and signaling via the inhibin-alpha subunit promoter in this study15 by removing the interaction with the basepairs of the minor groove. The p.(Arg92Trp) mutation is not allowed in the structure without gross structural rearrangements; the W side chain cannot insert into the minor groove and cannot be accommodated in the current DNA-bound structure without inducing steric clashes.
Transcriptional activity and subcellular localization of NR5A1 mutants p.(Arg92Trp) and p.(Arg92Gln)
We assessed the impact of following NR5A1 mutations on transcriptional activity and subcellular localization in different cellular systems: c.274C>T p.(Arg92Trp) and the previously reported adjacent mutation c.275G>A p.(Arg92Gln). For the latter mutation, partial loss of DNA-binding and transcriptional activity have previously been shown.24 We cotransfected WT or mutagenized NR5A1 plasmids with different NR5A1 responsive promoters (TESCO, SOX9, AMH, CYP11B1) in different cell lines (HeLa, KGN). These experiments were repeated with FOXL2 cotransfection because it was shown previously that FOXL2 inhibits NR5A1-mediated SOX9 expression during female development.25 The results of these assays were inconclusive, and only the results for the TESCO-luc constructs are shown (Supplementary Data online, Supplementary Figure S1 online). These experiments reproduced the previously described lower transcriptional activity of the c.275G>A p.(Arg92Gln) variant. The c.274C>T p.(Arg92Trp) variant, however, led to decreased activity in HeLa cells but not in KGN cells, showing that the transcriptional activity of a variant might be influenced by the cellular environment. Subcellular localization assays in HeLa cells showed nuclear expression without aggregate formation for both WT and mutant NR5A1 (Supplementary Data online, Supplementary Figure S2 online).
Transcriptome analysis in patients’ lymphocytes
RNA-seq generated an average of 39 million reads per sample. Differential expression analysis with default settings showed a total of 1,420 genes with significant differential expression (corrected P value < 0.05) when comparing the patient and control groups (Supplementary Data online, Supplementary Table S17 online). This gene set was filtered against a list of DSD-associated genes (Supplementary Data, Supplementary Table S2), the coverage of which was low in general. This filtering resulted in three (LEPR, MAMLD1, and IRF2BPL) genes with differential expression (Supplementary Data, Supplementary Table S18). MAMLD1, a direct target gene of NR5A1 (ref. 26) that was previously reported to be involved in 46,XY DSD (ref. 27), was upregulated (log 2-fold change = 1.637, P = 0.03) in patients compared with controls. BioGPS, an online available expression database, shows expression in both testis and ovaries in mice. LEPR and IRF2BPL (EAP1) are genes involved in hypogonadotropic hypogonadism, which we considered not directly relevant in the context of the phenotypes observed here.
Immunostaining of SOX9, FOXL2, and DDX4 on patients’ gonads
Gonadal biopsy specimens from cases 1 and 3 and a gonadectomy specimen from case 2 were available. In case 1, a dysgenetic testis with scarce germ cells, as identified by DDX4 immunostaining, was found. Biopsy specimens for case 3 revealed bilateral immature testes with Sertoli cell–only tubules (i.e., no germ cells present). Case 2 had bilateral ovotestes, including the presence of primordial and primary follicles in the ovarian part, located at the pole of the gonad, and next to immature seminiferous tubules without germ cells, located in the central testicular part of both gonads. Immunohistochemistry with SOX9 and FOXL2 antibodies, identifying Sertoli and granulosa cell differentiation, respectively, revealed exclusive SOX9 expression in testicular parts in all three cases and exclusive FOXL2 expression in ovarian parts of case 2 ( Figure 3 ). Hematoxylin and eosin staining of ovarian tissue, removed after ovarian torsion at age 41 years in the mother of case 2, was unremarkable and showed an aging ovary with some residual primordial follicles and stromal tissue (Supplementary Data online, Supplementary Figure S3 online).

Immunohistochemistry on gonadal biopsies. (a) Case 1. Testis. Supportive cells have differentiated as Sertoli cells as indicated by SOX9 positivity. SOX9 staining, 200×. (b) Case 1. Scarce germ cells (stained in brown) populate the testis tubules. DDX4 staining, 200×. (c) Case 2. Ovotestis. Sertoli cells in the testicular component (right) express SOX9; no SOX9 expression is seen in the ovarian part (left). SOX9 staining, 200×. (d) Same gonad. Ovarian granulosa cells but not testicular Sertoli cells display FOXL2 positivity. FOXL2 staining, 200×. (e,f) Germ cells (ova, black arrows) are only present in the ovarian and not in the testicular part. DDX4 staining, 200×. (g) Case 3. In this patient, bilateral infantile testes with a Sertoli cell–only pattern were observed. All Sertoli cells are SOX9-positive. SOX9 staining, 200×. (h) Same gonad. FOXL2 staining is negative. FOXL2, 200×. (i) Case 3. DDX4 staining indicates absence of germ cells, thus confirming the Sertoli cell–only pattern. DDX4 staining, 200×.
Discussion
A few NR5A1 mutations have been identified in females with 46,XY complete gonadal dysgenesis and adrenal failure.24 Interestingly, one of these was an adjacent homozygous mutation, c.275G>A p.(Arg92Gln), in the same codon as the c.274C>T p.(Arg92Trp) mutation identified here. Lower transcriptional activity was shown for p.(Arg92Gln), suggesting a loss-of-function effect.24 This mutation was also recently identified in a 46,XX female with adrenal insufficiency but no gonadal fenotype.28 Subsequently, numerous NR5A1 mutations and copy number variations have been associated with isolated 46,XY gonadal dysgenesis, 46,XY undervirilization, and male infertility.29,30,31,32 In 46,XX individuals, these loss-of-function mutations may cause primary ovarian insufficiency (POI).33 This broad range of phenotypes emphasizes that correct NR5A1 functioning is essential for both male and female gonadal development and maintenance.
Few data exist regarding the NR5A1 expression pattern in human female and male fetuses. In humans, NR5A1 is upregulated in the undifferentiated stage and gonadal expression is maintained during the entire period of fetal development in both male and female fetuses, whereas in rodents it has been shown that Nr5a1 expression decreases in the ovary as sex differentiation progresses.34,35 Indeed, NR5A1 plays an important role in multiple stages of gonadal development because it is a key regulator of sex determination via SOX9 upregulation during male development and of sex differentiation in both sexes via upregulation of AMH and the different steroidogenic enzymes.23,36,37 Both male and female Nr5a1 knockout (KO) mice lack gonadal and adrenal development.38,39 Recently, a CRISPR/Cas9 amino acid substituted mouse model was generated for p.(Arg92Trp); however, no phenotypic data were provided.40
Here, the NR5A1 mutation p.(Arg92Trp) was found in three unrelated patients with 46,XX (ovo)testicular DSD. A potential founder effect was suggested by haplotype analysis. The mutation was found in XX unaffected individuals in each of the three families, suggesting incomplete penetrance, as has been observed in other NR5A1-associated phenotypes41,42 and in other large families with 46,XX (ovo)testicular DSD.12,13 In a 46,XY DSD mouse model (B6 XYpos), incomplete penetrance has been shown to result from differences in spatiotemporal expression.43 Of note, the geographical distribution of testicular and ovarian regions in the ovotestes of case 2 supports the involvement of analogous (temporospatial) mechanisms and corresponds to observations in the B6 XYpos mouse.43
During normal ovarian development, FOXL2 actively represses SOX9, thereby inhibiting the male developmental cascade.44 During male development, it is well established that NR5A1, together with other transcription factors, binds to the regulatory domain of SOX9, thereby inducing a positive feedback loop in which SOX9 maintains and enhances its own expression, resulting in Sertoli cell recruitment and differentiation.36 NR5A1 loss-of-function mutations lead to a decrease in activation of SOX9 and other transcriptional targets, resulting in undervirilization and 46,XY DSD. Our initial hypothesis that p.(Arg92Trp) leads to more stable binding of NR5A1 to the SOX9 promoter, thereby directly inhibiting FOXL2-mediated repression of SOX9, as described by Uhlenhaut et al.,25 is not supported by the cellular transactivation assays performed. This is possibly because the adult cell lines that were used are not representative for early stages of gonadal development. In addition, transcriptome analysis of patient-derived lymphocytes did not reveal an upregulation of early male-specific genes. However, it did show upregulation of another NR5A1 target, MAMLD1. Whether this finding represents a direct effect resulting from p.(Arg92Trp) or whether it is secondary to the male environment from which cells were taken is currently unclear.
A more attractive hypothesis is that NR5A1, apart from its well-known role in inducing testicular development, also triggers the activation of early ovarian genes such as WNT4, a pathway that ultimately leads to FOXL2 activation and definitive suppression of the NR5A1-SOX9 tandem. Repression of the Nr5a1 cofactor Cited2 in XX mice entails reduced Nr5a1 activity, which has been shown to result in decreased Wnt4, Rspo1, and Foxl2 levels and increased mesonephric cell migration and Fgf9 levels, leading to transient activation of the male pathway in XX gonads.45 It is conceivable that in our patients, p.Arg92Trp interferes with this early ovary-promoting function of NR5A1, ultimately resulting in insufficient and/or delayed upregulation of FOXL2 and, thus, (partial) escape from its testis-suppressing activity. In our patients, NR5A1 is directly affected, which might explain the permanent and more severe gonadal phenotype as compared to the mouse model. Given the fact that the mutation affects the Ftz-F1 domain, which is important for DNA-binding specificity and stabilization, an altered expression of specific targets might be expected. In addition, this hypothesis fits with the incomplete penetrance and phenotypic heterogeneity, as seen in our families, depending on timing and spatial thresholds.
Two animal models support this hypothesis. It was shown very recently that CRISPR/Cas9-induced Nr5a1 mutations cause female-to-male sex reversal in the XX Nile Tilapia. Nr5a1 depletion leads to reduced Cyp19A1 expression and low estradiol levels. Because ovarian differentiation in fish is mediated by estradiol, the lack of sex steroid production here was proposed as the possible trigger for changing the initial gonadal fate.46 Temporal expression studies of Nr5a1 and Foxl2 in this model underscore the critical importance of threshold levels of Foxl2 to downregulate Nr5a1 in the XX gonad. A second animal model is the polled intersex syndrome (PIS) goat, which is characterized by absence of horns in heterozygous and homozygous animals and female-to-male sex reversal in homozygous XX animals. This phenotype is caused by the 11.7-kb regulatory deletion that is located 300 kb upstream of the goat FOXL2 gene. Knockout experiments have shown that FOXL2 depletion leads to reduced CYP19A1 expression and that it is responsible for inhibiting ovarian development47 similarly to the fish model. However, if testis formation in this species is, similarly to the Nile Tilapia, mediated by reduced Nr5a1 activity, then its influence on the female gonadal pathway remains to be elucidated.
These two alternative hypotheses may explain the different phenotypes observed for the two adjacent NR5A1 mutations. Mutation c.275G>A (p.Arg92Gln) has a direct negative influence on SOX9 expression and leads to aberrant male gonadal development and adrenal failure.24 Variant p.Arg92Trp has no direct effect on SOX9 expression but is hypothesized to interfere with hitherto unrecognized female-promoting activity of NR5A1, ultimately resulting in loss of stable SOX9 repression by FOXL2. Both mutations are located in the same codon that is part of the Ftz-F1 box, which is important for DNA-binding specificity. The newly introduced amino acid might determine which NR5A1 targets are affected by the mutation and which pathways are perturbed. Structural models show that both mutations have problems entering the DNA minor groove, although they cannot predict which targets will be affected. Our results suggest that temporal, spatial, or quantitative changes disturbing the balance between the male and female pathways in the XX gonad may shift the ovarian developmental program to the testicular pathway, probably through autocrine and paracrine reinforcing signals43 ( Figure 4 ). The exact mechanisms behind these processes need to be substantiated by additional experimental work.

Schematic overview of the sex development gene regulation network. Blue: testis-promoting activity of NR5A1 (ref. 1). Orange: counteractive connections to suppress the opposite pathway. Green: ovary-promoting activity of NR5A1 as hypothesized here and supported by other works.45,46 (a) General scheme: NR5A1 is known to initiate the male developmental pathway through upregulation of SOX9 (synergistically with SRY). SOX9 then maintains its own expression via Fgf9 signaling. In the female embryo, in the absence of SRY, NR5A1 induces WNT4 and RSPO1 expression as shown by Combes, leading to upregulation of FOXL2 and other ovary-specific genes. High FOXL2 expression results in stable repression of SOX9 and, possibly, NR5A1, and hence the male pathway. (b) Possible mechanism by which c.274C<T leads to ovotesticular disorders of sex development. We hypothesize that this novel mutation affects the activation of female-specific genes such as WNT4/β-catenin (as indicated by the dotted lines), leading to decreased FOXL2 expression. In this way, FOXL2 can no longer prosecute its pro-ovarian functions and, at the same time, male-promoting genes escape firm suppression, ultimately resulting in NR5A1-mediated and/or independent SOX9 upregulation and enhancement of testicular differentiation.
Conclusion
Taking these findings together, we propose NR5A1 as a novel disease gene for 46,XX (ovo)testicular DSD. Our study suggests SRY-independent SOX9 expression at the gonadal (testicular) level, most likely not through the canonical NR5A1-SOX9 interaction, but rather through downregulation of the pro-ovarian Wnt4/β-catenin pathways, thus tipping the balance toward male development. In addition, we demonstrated that testicular and ovotesticular DSD may represent different phenotypes resulting from a common cause. We conclude that NR5A1, a well-established gene for male gonadal development, is also involved in correct female gonadal development, the exact molecular mechanisms of which are still elusive.
Disclosure
The authors declare no conflict of interest.
References
Ono M, Harley VR. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol 2013;9:79–91.
Li TF, Wu QY, Zhang C, et al. 46,XX testicular disorder of sexual development with SRY-negative caused by some unidentified mechanisms: a case report and review of the literature. BMC Urol 2014;14:104.
Huang B, Wang S, Ning Y, Lamb AN, Bartley J. Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet 1999;87:349–353.
Benko S, Gordon CT, Mallet D, et al. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet 2011;48:825–830.
Sutton E, Hughes J, White S, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest 2011;121:328–341.
Haines B, Hughes J, Corbett M, et al. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J Clin Endocrinol Metab 2015;100:E815–E820.
Seeherunvong T, Perera EM, Bao Y, et al. 46,XX sex reversal with partial duplication of chromosome arm 22q. Am J Med Genet A 2004;127A:149–151.
Aleck KA, Argueso L, Stone J, Hackel JG, Erickson RP. True hermaphroditism with partial duplication of chromosome 22 and without SRY. Am J Med Genet 1999;85:2–4.
Polanco JC, Wilhelm D, Davidson TL, Knight D, Koopman P. Sox10 gain-of-function causes XX sex reversal in mice: implications for human 22q-linked disorders of sex development. Hum Mol Genet 2010;19:506–516.
Parma P, Radi O, Vidal V, et al. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet 2006;38:1304–1309.
Tomaselli S, Megiorni F, De Bernardo C, et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat 2008;29:220–226.
Slaney SF, Chalmers IJ, Affara NA, Chitty LS. An autosomal or X linked mutation results in true hermaphrodites and 46,XX males in the same family. J Med Genet 1998;35:17–22.
Kuhnle U, Schwarz HP, Löhrs U, Stengel-Ruthkowski S, Cleve H, Braun A. Familial true hermaphroditism: paternal and maternal transmission of true hermaphroditism (46,XX) and XX maleness in the absence of Y-chromosomal sequences. Hum Genet 1993;92:571–576.
Ramos ES, Moreira-Filho CA, Vicente YA, et al. SRY-negative true hermaphrodites and an XX male in two generations of the same family. Hum Genet 1996;97:596–598.
Little TH, Zhang Y, Matulis CK, et al. Sequence-specific deoxyribonucleic acid (DNA) recognition by steroidogenic factor 1: a helix at the carboxy terminus of the DNA binding domain is necessary for complex stability. Mol Endocrinol 2006;20:831–843.
Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 2002;320:369–387.
Krieger E, Joo K, Lee J, et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct Funct Bioinforma 2009;77:114–122.
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–1612.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–1111.
Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–515.
Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2014;31:0–5.
Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47.
Parker KL, Schimmer BP. Steroidogenic factor 1: a key determinant of endocrine development and function. Endocr Rev 1997;18:361–377.
Achermann J, Ozisik G, Ito M, et al. Gonadal determination and adrenal development are steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab 1999;87:1829–1833.
Uhlenhaut NH, Jakob S, Anlag K, et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 2009;139:1130–1142.
Fukami M, Wada Y, Okada M, et al. Mastermind-like domain-containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J Biol Chem 2008;283:5525–5532.
Kalfa N, Fukami M, Philibert P, et al. Screening of MAMLD1 mutations in 70 children with 46,XY DSD: identification and functional analysis of two new mutations. PLoS One 2012;7:e32505.
Guran T, Buonocore F, Saka N, et al. Rare causes of primary adrenal insufficiency: genetic and clinical characterization of a large nationwide cohort. J Clin Endocrinol Metab 2016;101:284–292.
Köhler B, Lin L, Ferraz-de-Souza B, et al. Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46,XY patients with severe underandrogenization but without adrenal insufficiency. Hum Mutat 2008;29:59–64.
Barbaro M, Cools M, Looijenga LH, Drop SL, Wedell A. Partial deletion of the NR5A1 (SF1) gene detected by synthetic probe MLPA in a patient with XY gonadal disorder of sex development. Sex Dev 2011;5:181–187.
Bashamboo A, Ferraz-de-Souza B, Lourenço D, et al. Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am J Hum Genet 2010;87:505–512.
Köhler B, Lin L, Mazen I, et al. The spectrum of phenotypes associated with mutations in steroidogenic factor 1 (SF-1, NR5A1, Ad4BP) includes severe penoscrotal hypospadias in 46,XY males without adrenal insufficiency. Eur J Endocrinol 2009;161:237–242.
Lourenço D, Brauner R, Lin L, et al. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med 2009;360:1200–1210.
Hanley NA, Ball SG, Clement-Jones M, et al. Expression of steroidogenic factor 1 and Wilms’ tumour 1 during early human gonadal development and sex determination. Mech Dev 1999;87:175–180.
Cools M, Hoebeke P, Wolffenbuttel KP, et al. Pubertal androgenization and gonadal histology in two 46,XY adolescents with NR5A1 mutations and predominantly female phenotype at birth. Eur J Endocrinol 2012;166:341–349.
Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 2008;453:930–934.
Shen WH, Moore CC, Ikeda Y, Parker KL, Ingraham HA. Nuclear receptor steroidogenic factor 1 regulates the müllerian inhibiting substance gene: a link to the sex determination cascade. Cell 1994;77:651–661.
Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994;77:481–490.
Sadovsky Y, Crawford PA, Woodson KG, et al. Mice deficient in the orphan receptor steroidogenic factor 1 lack adrenal glands and gonads but express P450 side-chain-cleavage enzyme in the placenta and have normal embryonic serum levels of corticosteroids. Proc Natl Acad Sci USA 1995;92:10939–10943.
Inui M, Miyado M, Igarashi M, et al. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci Rep 2014;4:5396.
Camats N, Pandey AV, Fernández-Cancio M, et al. Ten novel mutations in the NR5A1 gene cause disordered sex development in 46,XY and ovarian insufficiency in 46,XX individuals. J Clin Endocrinol Metab 2012;97:E1294–E1306.
Baetens D, Mladenov W, Delle Chiaie B, et al. Extensive clinical, hormonal and genetic screening in a large consecutive series of 46,XY neonates and infants with atypical sexual development. Orphanet J Rare Dis 2014;9:209.
Wilhelm D, Washburn LL, Truong V, Fellous M, Eicher EM, Koopman P. Antagonism of the testis- and ovary-determining pathways during ovotestis development in mice. Mech Dev 2009;126:324–336.
Veitia RA. FOXL2 versus SOX9: a lifelong “battle of the sexes”. Bioessays 2010;32:375–380.
Combes AN, Spiller CM, Harley VR, et al. Gonadal defects in Cited2-mutant mice indicate a role for SF1 in both testis and ovary differentiation. Int J Dev Biol 2010;54:683–689.
Xie QP, He X, Sui YN, Chen LL, Sun LN, Wang DS. Haploinsufficiency of SF-1 causes female to male sex reversal in Nile Tilapia, Oreochromis niloticus. Endocrinology 2016;157:2500–2514.
Boulanger L, Pannetier M, Gall L, et al. FOXL2 is a female sex-determining gene in the goat. Curr Biol 2014;24:404–408.
Acknowledgements
We are very grateful to the families who participated in this study. This work was supported by a grant from the Ghent University Special Research Fund (BOF15/GOA/011) to E.D.B., by Belspo IAP project P7/43 (Belgian Medical Genomics Initiative: BeMGI) to E.D.B., by the Hercules Foundation (AUGE/13/023 to E.D.B.), by a grant from the Ghent University Special Research Fund (BOF Starting Grant) to M.C., and by grant G0D6713N from the Research Foundation Flanders (FWO) to E.D.B. and M.C. E.D.B. and M.C. are Senior Clinical Investigators of the FWO. F.C. is senior postdoctoral fellow of the FWO. We thank Francis Poulat for providing the TESCO-luc construct and Jan Tavernier for providing the NR5A1 ORF.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Data
(ZIP 4012 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Baetens, D., Stoop, H., Peelman, F. et al. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet Med 19, 367–376 (2017). https://doi.org/10.1038/gim.2016.118
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.118
Keywords
This article is cited by
-
Evidence for NR2F2/COUP-TFII involvement in human testis development
Scientific Reports (2024)
-
Genetic control of typical and atypical sex development
Nature Reviews Urology (2023)
-
Characteristics and possible mechanisms of 46, XY differences in sex development caused by novel compound variants in NR5A1 and MAP3K1
Orphanet Journal of Rare Diseases (2021)
-
Cellular fate of intersex differentiation
Cell Death & Disease (2021)
-
Towards improved genetic diagnosis of human differences of sex development
Nature Reviews Genetics (2021)
