
Abstract
The combination of both very high brightness and deep blue emission from phosphorescent organic light-emitting diodes (PHOLED) is required for both display and lighting applications, yet so far has not been reported. A source of this difficulty is the absence of electron/exciton blocking layers (EBL) that are compatible with the high triplet energy of the deep blue dopant and the high frontier orbital energies of hosts needed to transport charge. Here, we show that N-heterocyclic carbene (NHC) Ir(III) complexes can serve as both deep blue emitters and efficient hole-conducting EBLs. The NHC EBLs enable very high brightness (>7,800 cd m−2) operation, while achieving deep blue emission with colour coordinates of [0.16, 0.09], suitable for most demanding display applications. We find that both the facial and the meridional isomers of the dopant have high efficiencies that arise from the unusual properties of the NHC ligand—that is, the complexes possess a strong metal–ligand bond that destabilizes the non-radiative metal-centred ligand-field states. Our results represent an advance in blue-emitting PHOLED architectures and materials combinations that meet the requirements of many critical illumination applications.
Similar content being viewed by others

Main
A driving force behind the use of organic electroluminescence in displays and lighting has been the introduction of red and green electrophosphorescent devices with up to 100% internal quantum efficiencies1,2. However, achieving deep blue electrophosphorescence with high efficiency and brightness, together with long-term operational stability, remains a significant challenge3. The design of robust and efficient blue phosphors free of electrochemically reactive moieties offers one possible solution. For example, saturated blue-emitting tris-cyclometalated iridium(III) complexes4,5 were introduced by using the thermodynamically stable N-heterocyclic carbene (NHC) ligands, or Ir(C∧C:)3 (ref. 6). This is compared with blue Ir complexes using fluorination to obtain a wide energy gap7,8, which has resulted in high efficiency but only an unsaturated sky-blue colour, unsuitable for displays. Unfortunately, a significant drawback of previously demonstrated deep blue phosphorescent organic light-emitting diodes (PHOLEDs) is that they are subject to a pronounced efficiency roll-off at the high brightness required in most display and lighting applications7,8,9,10,11,12,13,14,15. For example, the current density at the half-maximum EQE, J1/2, is typically <50 mA cm−2 owing to the loss of excitons and electrons from the PHOLED emission layer (EML), as well as strong bimolecular annihilation16. Thus, they barely achieve a brightness >3,000 cd m−2 at J1/2. Up to the present work, preventing these parasitic effects has been exacerbated by the lack of exciton and charge blocking layers that are compatible with the high-energy triplet emitters required for deep blue emission.
Based on our understanding of the unique energetics of the Ir(C∧C:)3 complexes, we introduce a device design that significantly improves the efficiency of the deep blue PHOLEDs, especially at high brightness. Here, Ir(C∧C:)3 complexes are used as the phosphorescent emitting molecules, electron/exciton blocking layers (EBL) and dopants that conduct holes across the EML. The combined effects of these multiple uses lead to a marked improvement in EQE at high current densities. Specifically, J1/2 is increased in devices with an EBL by more than a factor of 280 compared to those without it, and is improved by a further 50% (resulting in a cumulative improvement by a factor of 420) by grading the dopant across the EML, thereby reducing triplet annihilation losses at very high brightness17,18.
Facial (fac-) and meridional (mer-)Ir(C∧C:)3-based PHOLEDs exhibit Commission Internationale d’Eclairage (CIE) coordinates of [0.16, 0.09] and [0.16, 0.15], with maximum external quantum efficiencies of EQE = 10.1 ± 0.2 and 14.4 ± 0.4% at low luminance, decreasing slightly to 9.0 ± 0.1 and 13.3 ± 0.1% at L = 1,000 cd m−2. Surprisingly, the device efficiencies are decreased by 50% at unusually high brightness, giving L = 7,800 ± 400 and 22,000 ± 1,000 cd m−2 (corresponding to J1/2 = 160 ± 10 and 210 ± 1 mA cm−2), respectively. The fac- or mer-Ir(C∧C:)3-based devices produce unparalleled brightness at J1/2 compared with the PHOLEDs with similar colour coordinates15,19. To our knowledge, the fac-isomer-based device achieves the brightest deep blue emission among the PHOLEDs reported so far, which nearly meets the most stringent National Television System Committee (NTSC) requirements.
An additional finding is that the mer-Ir(C∧C:)3 is equally or more efficiently luminescent than the fac-isomer in solution and the solid state20, whereas conventional red- and green-emitting Ir(C∧N)3 complexes follow the opposite trend—that is, fac is more efficient than mer21,22. We find that the strong Ir–NHC ligand bonds4 in Ir(C∧C:)3 result in reduced non-radiative decay via the metal-centred ligand-field states for both isomers23. Our studies of the photophysics of Ir(C∧C:)3 complexes, along with their employment in device architectures designed for their optimum performance, provides a solution for achieving efficient deep blue emission at very high brightness.
Results
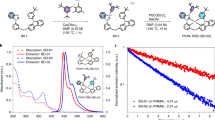
The structure of our newly synthesized NHC Ir(III) complex, tris-(N-phenyl, N-methyl-pyridoimidazol-2-yl)iridium(III), Ir(pmp)3, is based on the near-ultraviolet-emitting tris-(N-phenyl, N-methyl-benzimidazol-2-yl) Ir(III), or Ir(pmb)3, whose benzannulated component in the NHC ligands is replaced with a fused pyridyl ring, as shown in Fig. 1a. The greater electronegativity of the nitrogen atom versus methine (CH) lowers the reduction potential of fac-Ir(pmp)3 to Ered = −2.77 ± 0.05 V relative to fac-Ir(pmb)3 (Ered = −3.19 ± 0.05 V), although their oxidation potentials are nearly identical (Eox = 0.47 ± 0.05 and 0.45 ± 0.05 V, respectively). The absorption spectra of fac-Ir(pmb)3 and fac-Ir(pmp)3 in Fig. 1b show that their spin-allowed metal-to-ligand charge-transfer (1MLCT) transitions have high-energy onsets, at λ = 350 and 380 nm, respectively. The observed redshift for the absorption spectrum of fac-Ir(pmp)3 results from its smaller energy gap compared to fac-Ir(pmb)3 inferred from their Ered and Eox. Accordingly, the photoluminescence (PL) spectrum of the fac-Ir(pmp)3 in Fig. 1c has a redshifted peak wavelength of λmax = 418 nm in the deep blue compared to the near-ultraviolet emission of fac-Ir(pmb)3 (λmax = 380 nm; ref. 4). Meanwhile, the PL spectrum of mer-Ir(pmp)3 is broad and exhibits a large room-temperature bathochromic shift (λmax = 465 nm) relative to the fac-isomer. The lower emission energy of the mer-Ir(pmp)3 compared to the fac-isomer is due to its lower oxidation potential (Eox = 0.23 ± 0.05 V) and nearly identical reduction potentials (Ered = −2.80 ± 0.05 V), which result in a correspondingly reduced energy gap. The emission from both Ir(pmp)3 isomers undergoes a pronounced rigidochromic shift at T = 77 K, with the fac-isomer exhibiting a vibronically structured line shape.

a, Molecular structural formulae of fac-Ir(pmb)3 and the fac- and mer-isomers of Ir(pmp)3. Here, fac- and mer-Ir(pmp)3 have the C3 andC1 molecular symmetries, respectively, in pseudo-octahedral coordinates. b, Absorption spectra of fac-Ir(pmb)3 (circles), fac-Ir(pmp)3 (squares) and mer-Ir(pmp)3 (triangles) in dichloromethane (CH2Cl2) solution. c, Photoluminescence spectra of fac- and mer-Ir(pmp)3 in degassed 2-methyltetrahydrofuran (2-MeTHF) at temperatures of T = 295 K and 77 K.
Figure 2a shows the temperature-dependent transient PL characteristics of fac- and mer-Ir(pmp)3 in de-aerated 2-MeTHF, with quantum yields of ΦPL = 76 ± 5 and 78 ± 5%, respectively, at T = 295 K (compare with ΦPL of fac-Ir(pmb)3 = 37 ± 5%; ref. 23) and ΦPL = 95 ± 5% at 77 K for both isomers. The triplet lifetimes, τ, were obtained from mono-exponential fits to the data at room temperature. Radiative (kr) and non-radiative (knr) rate constants are calculated using the relationship22kr = ΦPL/τ, where ΦPL = kr/(kr + knr). The mer-isomer has a shorter triplet lifetime of τ = 0.8 ± 0.1, versus 1.2 ± 0.1 μs for the fac-isomer, which results in its higher kr = (1.0 ± 0.2) × 106, versus (6.4 ± 1.3) × 105 s−1, and knr = (2.7 ± 0.4) × 105, versus (2.0 ± 0.4) × 105 s−1. At T = 77 K, triplet lifetimes of fac-Ir(pmp)3 were extracted from multi-exponential fits. Accordingly, fac-Ir(pmp)3 has two relatively well-resolved lifetimes of τ1 = 3.9 ± 0.2 μs (weighting: 45%) and τ2 = 9.2 ± 0.2 μs (55%). In contrast, the mer-isomer still shows a mono-exponential decay, with an only slightly increased τ = 1.0 ± 0.1 μs at T = 77 K. Table 1 summarizes the photophysical parameters of both isomers.

a, Transient phosphorescence decay of diluted fac- and mer-Ir(pmp)3 in 2-MeTHF obtained at T = 295 and 77 K, with the fits based on mono- or multi-exponential decays. b, Photoluminescence spectra of diluted fac- and mer-Ir(pmp)3 in different polarity media, poly(methyl methacrylate) (PMMA), toluene and dichloromethane (DCM).
Figure 2b shows the PL spectra of diluted fac- and mer-Ir(pmp)3 in media of different polarity, poly(methyl methacrylate) (PMMA), toluene and dichloromethane (DCM), having dipole moments of 0, 0.42 and 1.6 D (ref. 24), respectively. Both complexes exhibit stronger positive solvatochromism in a more polar medium. Further, the PL spectrum of the mer-isomer is more redshifted and broadened than that of the fac-isomer, because the excited states of the former complex are more stabilized in a similar polar solvent.
Figure 3 shows the structures of PHOLEDs using fac- or mer-Ir(pmp)3 (denoted by devices Dfac or Dmer) along with the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies for all organic materials studied25,26,27. The LUMO energies are calculated from the reported or measured Ered (following ref. 27). The EBLs consist of the Ir(C∧C:)3 themselves (that is, fac-Ir(pmp)3 and fac-Ir(pmb)3 for Dfac and Dmer, respectively), which have equal or shallower LUMO energies than that of the host, as well as equal or larger triplet energy levels than the dopants. This enables efficient hole injection into the hole-conducting dopants, while blocking electrons transported via the host. The doping concentration in the EML is linearly graded from 20 vol% at the EBL interface to 8 vol% at the hole blocking layer (HBL) interface to create a uniform triplet distribution across the EML (ref. 17).

a,b, Energy level diagrams of phosphorescent organic light-emitting devices (PHOLEDs) based on fac- (a) and mer-Ir(pmp)3 (b), denoted by devices Dfac and Dmer, respectively. Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies of comprising materials in eV are either calculated or obtained from the literature26,27,28. HIL, hole injection layer; HTL, hole blocking layer; EBL, electron/exciton blocking layer; EML, emissive layer; HBL, hole/exciton blocking layer; ETL, electron-transport layer; the corresponding number below each abbreviation is the thickness of the layer in nanometres. CzSi stands for 9-(4-tert-butylphenyl)-3,6-bis(triphenylsilyl)-9H-carbazole and TPBi stands for 1,3,5-tris(1-phenyl-1H-benzimidazol-2-yl)benzene.
Holes and electrons injected into the EML are mainly transported via the dopant and the host (diphenyl-4-triphenylsilylphenyl-phosphine oxide, or TSPO1), respectively (see Supplementary Section 1). Owing to the nested HOMO and LUMO energies of Ir(pmp)3 in TSPO1 (Fig. 3), the majority of electrons transported via the host are trapped by the dopant and then radiatively recombine with the holes on the dopant. As TSPO1 is preferably electron-transporting owing to its diphenylphosphine oxide group28, triplets are primarily formed at the EBL/EML interface (top, Fig. 4a). This necessitates the use of EBLs with high triplet and shallow LUMO energies, as shown in Fig. 3. In the graded EML, an initially high doping concentration (20 vol%) near the EBL/EML interface facilitates hole injection and transport, which gradually reduces owing to the decreasing dopant fraction (8 vol%) at the EML/HBL interface (bottom, Fig. 4a). The resulting triplet densities17 (or recombination profiles) of both types of EML are shown in Fig. 4b (see Methods). In the uniformly doped EML, ∼47% of triplets are concentrated near the EBL/EML interface (x = 0–10 nm), which decreases to approximately 33% in the graded EML owing to the deeper hole penetration. The distributed recombination profile reduces the probability of bimolecular annihilation quenching16. In addition, radiative recombination in the graded-EML device occurs farther from the EBL/EML interface compared to the uniformly doped EML PHOLED, resulting in enhanced light outcoupling (see Supplementary Section 2). These combined effects lead to the increased EQE of Dmer, as shown in the inset of Fig. 4b.

a, Charge-transport mechanisms in the emission layers (EML) of uniformly doped Ir(pmp)3 (blue circle) at 14 vol% (top) and linearly graded doping at 20–8 vol% (bottom) into TSPO1 (white rectangular background), from the EBL to the HBL boundaries. Black and red arrows describe the hole- and electron-transport trajectories, respectively, which then recombine radiatively, as illustrated by a yellow starburst. This conceptually demonstrates that recombination occurs relatively closer to the HBL boundary for the graded versus uniformly doped EML owing to the improved hole injection and transport of the former. b, Triplet density distributions of the uniformly doped (squares) and graded doped (circle) EMLs in Dmer measured at J = 10 mA cm−2. The external quantum efficiency (EQE) versus J of Dmer for both types of EML structure is compared in the inset.
Figure 5a shows the PL spectra of fac- and mer-Ir(pmp)3 uniformly doped at 14 vol% into TSPO1. Figure 5b shows the electroluminescence (EL) spectra of Dfac and Dmer measured at a current density of J = 10 mA cm−2, which result in deep blue CIE coordinates of [0.16, 0.09] and [0.16, 0.15], respectively. In the inset we show images of the packaged devices along with their characteristic emission colour. The EL spectra have nearly identical CIE coordinates to the PL, confirming the emission is solely from the dopants in the PHOLEDs. Figure 5c shows the current density–voltage–luminance (J–V–L) characteristics of Dfac and Dmer. Dmer turns on at a lower voltage than Dfac (3 V versus 4 V), which is presumably due to different charge injection, transport and trapping characteristics20, and the lower HOMO energy of mer-Ir(pmp)3 than the fac-isomer. Although the current densities of Dfac at high voltage (>7 V) are greater than those of Dmer, the latter device still achieves a higher luminance owing to its redshifted emission and higher EQE at all current densities.

a, Photoluminescence (PL) spectra of fac- and mer-Ir(pmp)3 doped at 14 vol% into TSPO1 excited by a HeCd laser (wavelength, λ = 325 nm). b, Electroluminescence (EL) spectra of Dfac and Dmer measured at a current density of J = 10 mA cm−2. The insets are photographs of 2 mm2 packaged PHOLEDs whose illumination is reflected from a white background to avoid saturation of the camera sensor. The bright irregular square shape in each image is due to light scattered from the epoxy package seal. The package is a sandwich of two glass slides, one containing electrodes (dark regions) and the other serving as a lid. c, Current density–voltage–luminance (J–V–L) characteristics of Dfac and Dmer. d, EQE versus J for PHOLEDs of either linearly graded or uniformly doped emission layers consisting of either fac- or mer-Ir(pmp)3. Fits (solid lines) are based on the model in the Methods. EQE versus J for the mer-based PHOLED without an electron/exciton blocking layer (EBL) is also plotted.
Figure 5d shows the EQE–J characteristics of Dfac, Dmer and analogous devices whose EMLs are uniformly doped at 14 vol% by fac and mer-Ir(pmp)3, and a device without an Ir(C∧C:)3-based EBL. By employing fac-Ir(pmb)3 as the EBL, the uniformly doped EML PHOLED has a markedly higher EQE and reduced efficiency roll-off at high J compared to the PHOLED lacking an EBL. Therefore, J1/2 increases by almost 280 times, from 0.5 ± 0.1 to 140 ± 10 mA cm−2, and EQE increases by at least 40% at all current densities. The EQEs of the graded-EML devices employing an EBL are further improved by ∼10% at all current densities compared to uniformly doped EML PHOLEDs, and J1/2 is increased by a further 50%, leading to a cumulative improvement by a significant factor of 420. Thus, Dfac and Dmer attain EQE = 10.1 ± 0.2 and 14.4 ± 0.4% at low luminance, decreasing only slightly to 9.0 ± 0.1 and 13.3 ± 0.1% at L = 1,000 cd m−2, and by 50% at L = 7,800 ± 400 and 22,000 ± 1,000 cd m−2 (corresponding to J1/2 = 160 ± 10 and 210 ± 10 mA cm−2), respectively. The difference in EQE of Dfac versus Dmer is consistent with the trend found in the solid-state PL quantum yields for fac- versus mer-Ir(pmp)3 when doped in TSPO1 at the same concentrations (2–30 vol%, see Supplementary Section 3).
The EQE–J plot of graded and uniformly doped fac- and mer-Ir(pmp)3 PHOLEDs in Fig. 5d was modelled to analyse the effect of the distributed recombination profile on device performance (see Methods and Table 2). As the doping concentration changes from uniform (14 vol%) to graded (20–8 vol%), the triplet–triplet annihilation rate (kTT) increases whereas the triplet–polaron annihilation rate (kTP) decreases for both fac- and mer-Ir(pmp)3-based devices. The increased kTT is due to a high triplet concentration in the dopant-rich (>14 vol%) region of the EML (Fig. 5b). However, the reduced local density of triplets in the graded EML compensates for the higher kTT. Also, the significantly lower kTP for the graded devices, which is proportional to the charge density29, is consistent with the measured profiles.
Discussion
Implication of the employed device design.
The graded deep-blue-emitting phosphor Ir(pmp)3 across the EML serves as a wide-energy-gap hole transporter that evenly distributes the exciton formation zone, thereby reducing bimolecular triplet annihilation. Note that light-blue-emitting materials such as iridium(III)bis[(4,6-difluorophenyl)-pyridinato-N,C2′]picolinate (FIrpic) achieve higher emission efficiency (>20%; refs 30,31) than reported here. This is a result of their relatively low emission energies and the large choice of host materials. Given the difficulties of synthesizing hole-transporting hosts for deep blue PHOLEDs, using hole-transporting gradient doping via the phosphor itself is clearly an effective strategy for improving efficiency. Although co-host systems using two different compounds to separately transport holes and electrons have previously been shown to improve charge balance in the EML (ref. 31), they do not generally eliminate the need for an EBL and/or HBL, as charge carriers and excitons can still leak out from the emission zone without them. To prevent leakage, the comparatively high electron mobility in the recent deep blue OLED EML (refs 15,32) necessitates using an EBL, which has minimal impact on the conventional hole-transport dominated structures33. The significantly improved EQE and J1/2 of our PHOLED primarily results from the Ir(C∧C:)3 EBL (Fig. 5d). As noted, effective blocking by the dopant itself is a unique property of the Ir(C∧C:)3 family of phosphors, with their very shallow LUMO energies and wide HOMO–LUMO gaps. Indeed, previously reported deep blue PHOLEDs lacking the EBL are similar to that of our unblocked device with its severe efficiency roll-off, whereas the blocked PHOLED characteristics are similar to those of conventional red- and green-emitting devices. This indicates the achievement of both charge and exciton confinement in the EML.
Origin of both highly emissive fac- and mer-Ir(pmp)3.
The mer-isomers of the conventional red- and green-emitting Ir(C∧N)3 complexes21,22 typically have a non-radiative decay rate (knr) at least an order of magnitude larger than their fac-isomers. This difference is attributed to a more efficient thermal population of non-radiative triplet metal-centred (3MC) ligand-field states that comprise Ir–ligand antibonding orbitals in the mer-isomer23,34. The asymmetric molecular structure (C1) of mer-isomers leads to trans-disposed Ir–N linkages that are more labile compared to the three equivalent Ir–N bonds in the C3-symmetric fac-isomers35. Therefore, the 3MC states of the mer-isomer are stabilized and thermally accessible compared to the fac-isomer. However, for Ir(pmp)3, the difference in knr between fac- and mer-isomers is less than a factor of two as a result of the strong Ir–carbene bonds destabilizing the 3MC states for both isomers. The lack of mer-to-fac isomerization for Ir(pmp)3 substantiates its strong metal–ligand bond nature, whereas the Ir(C∧N)3 complexes, having weaker Ir–N bonds, allow such conversion23. At T = 77 K, the quantum yields of fac- and mer-Ir(pmp)3 increase to near unity owing to suppressed non-radiative decay via a thermal population to 3MC states. The relative dominance of ligand-centred (3LC) over 3MLCT excited states in fac-Ir(pmp)3 compared to the mer-isomer is reflected in the more pronounced temperature dependence of the transient PL response (Fig. 2a). The broader PL spectrum, both at T = 295 and 77 K, more pronounced bathochromic shift in a polar medium (Fig. 2b), and rigidochromic shift in a frozen media (see Fig. 1c) confirm that emission from mer-Ir(pmp)3 originates more from a polar excited state (3MLCT), rather than the relatively nonpolar 3LC-dominant states of the fac-isomer36 (Supplementary Section 4). Compared to the structurally analogous violet-emitting fac-Ir(pmb)3 complex, fac- and mer-Ir(pmp)3 achieve a higher phosphorescence efficiency of ΦPL = 76 ± 5 and 78 ± 5%, respectively, relative to 37 ± 5% for fac-Ir(pmb)3 (ref. 23); the difference once more is due to the increased stabilization of the triplet states in Ir(pmp)3. Another possible explanation for the enhanced ΦPL is a decrease in the torsion angle between the phenyl and pyridoimidazole groups in Ir(pmp)3 relative to Ir(pmb)3, caused by a steric interference between the H-atoms at the 1,7 phenyl and benzimidazole group positions. Substitution of the methine (CH) for N in Ir(pmp)3 eliminates this conflict.
Differences between Dfac and Dmer.
The difference in the solid-state PL efficiencies of the fac- versus mer-Ir(pmp)3, which leads to the difference in EQE of Dfac versus Dmer, is probably due to different degrees of the emitter aggregation quenching. This may be induced by their different static dipole moments (17.2 D versus 10.8 D for fac- and mer-Ir(pmp)3, respectively), as high dipole moments can promote aggregation37. Hence, when fac- or mer-Ir(pmp)3 is doped at the same concentration of 11 vol% into wide-energy-gap hosts, the latter complex achieves higher PL quantum efficiencies (Supplementary Section 3). A polar host such as TSPO1 is comprised of phosphine oxide moieties (P = O), which may lead to further phosphorescence losses owing to crystallite formation38. That is, strong interactions between TSPO1 molecules lead to crystalline domains within an amorphous film that result in structured, lower-energy emission bands28 superposed with those obtained in solution39 (Supplementary Section 3). We speculate, therefore, that quenching by TSPO1 crystalline domains results in two phenomena: the lower PL efficiencies of Ir(pmp)3 in TSPO1 versus p-bis(triphenylsilyly) benzene (UGH2; ΦPL = 66 ± 3 versus 85 ± 4% for mer-Ir(pmp)3 at 11 vol%) despite similarly high triplet energies26 (ET = 3.37 and 3.5 eV, respectively) and reduced PL quantum yields of Ir(pmp)3 in TSPO1 at lower concentrations than those obtained at the optimal concentration of 14 vol%. The latter is in contrast to the results found with the conventional red, green and light-blue dopants into nonpolar hosts having maximum efficiency at very diluted concentrations (<2 vol%; ref. 40). On the other hand, the polar TSPO1 enables a high solid-state solubility of the Ir(pmp)3 by preventing interactions between dopants, leading to their physical separation28,41. This enables the comparatively high optimal doping concentration of 14 vol% of Ir(pmp)3, as well as a narrower and less bathochromically shifted Ir(pmp)3 emission, resulting in a more saturated blue emission compared with that in other nonpolar hosts. Finally, given that the PL quantum yields at 14 vol% in TSPO1 are higher than at either 20 vol% or 8 vol% of Ir(pmp)3 (the initial and terminal concentrations of the graded doping in the EML), the optimal doping concentrations for EL and PL are different because the dopant also serves as a hole transporter in the PHOLED, affecting the charge balance, and hence the EQE.
Conclusions
We find that deep-blue-emitting Ir(C∧C:)3 complexes can be simultaneously employed as triplet-emitting dopants, hole transporters and EBLs. This combination of uses significantly reduces electron and exciton losses. In particular, fac-Ir(pmp)3-based PHOLEDs achieved remarkably reduced efficiency roll-off at high current density, resulting in very high brightness (>7,800 cd m−2) with CIE coordinates of [0.16, 0.09], closest to the NTSC requirement among reported Ir-based PHOLEDs. The highly emissive mer-isomer of Ir(pmp)3, which is due to the strong Ir–NHC ligand bond, enables even brighter PHOLED (>22,000 cd m−2) operation in the blue. Our advances in materials design and device architectures provide the guidelines for designing efficient and, more importantly, long-lived deep blue PHOLEDs.
Methods
Synthesis of fac- and mer-isomers of Ir(pmp)3.
A mixture of N3-methyl-N2-phenylpyridine-2,3-diamine42 (4.11 g, 20.63 mmol), triethyl orthoformate (172 ml), concentrated HCl (0.9 ml), and 5 drops of formic acid was heated at reflux for 12 h under N2. The solution was cooled to room temperature, and the resulting solid (imidazolium chloride hydrochloric salt) was collected by filtration (2.81 g, 46%). A mixture of [IrCl(COD)]2 (700 mg, 1.04 mmol), ligand (1.76 g, 6.24 mmol), silver oxide (1.45 g, 6.24 mmol), and triethylamine (635 mg, 6.24 mmol) in chlorobenzene (100 ml) was bubbled with N2 for 10 min. The solution was heated to reflux for 12 h under N2. The reaction mixture was cooled and chlorobenzene was evaporated. The crude solid was coated on silica and twice purified by column chromatography [dichloromethane (DCM) to DCM/acetone = 95/5] to yield fac-Ir(pmp)3 (310 mg, 18%) and mer-Ir(pmp)3 (890 mg, 53%). CHN analyses were carried out on both compounds; theoretical: C 57.34, H 3.70, N 15.43; mer-Ir(pmp)3: C 57.22, H 3.52, N 15.52; fac-Ir(pmp)3: C 56.95, H 3.73, N 15.50. 1H and 13C nuclear magnetic resonance (NMR) data for the two complexes are given as follows; fac-Ir(pmp)3: 1H NMR (CDCl3, 400 MHz) d 8.84 (dd, J = 8.0, 1.2 Hz, 3H), 8.37 (dd, J = 8.0, 1.2 Hz, 3H), 7.39 (dd, J = 8.0, 1.2 Hz, 3H), 7.14–7.07 (m, 6H), 6.76 (td, J = 8.0, 1.2 Hz, 3H), 6.61 (dd, J = 8.0, 1.2 Hz, 3H), 3.28 (s, 9H); 13C NMR (CDCl3, 101 MHz) d 191.74, 147.61, 146.59, 146.25, 142.83, 136.44, 128.69, 125.36, 121.49, 116.89, 115.97, 114.68, 33.67; HRMS (m/z, ESI+) Calcd for C39H31IrN9 818.2332 [M + H+], found: 818.2347. mer-Ir(pmp)3:1H NMR (CDCl3, 400 MHz) d 8.88 (dd, J = 8.0, 1.2 Hz, 1H), 8.82 (dd, J = 8.0, 1.2 Hz, 1H), 8.78 (dd, J = 8.0, 1.2 Hz, 1H), 8.42 (dd, J = 4.8, 1.2 Hz, 2H), 8.38 (dd, J = 4.8, 1.2 Hz, 1H), 7.52 (dd, J = 8.0, 1.2 Hz, 1H), 7.49 (dd, J = 8.0, 1.2 Hz, 1H), 7.45 (dd, J = 8.0, 1.2 Hz, 1H), 7.22–7.13 (m, 3H), 7.07–6.97 (m, 3H), 6.93 (dd, J = 8.0, 1.2 Hz, 1H), 6.87 (dd, J = 8.0, 1.2 Hz, 1H), 6.76 (td, J = 8.0, 1.2 Hz, 1H), 6.70 (td, J = 8.0, 1.2 Hz, 1H), 6.66 (td, J = 8.0, 1.2 Hz, 1H), 6.56 (dd, J = 8.0, 1.2 Hz, 1H), 3.32 (s, 3H), 3.27 (s, 3H), 3.21 (s, 3H); 13C NMR (CDCl3, 101 MHz) d 190.48, 187.76, 186.63, 148.74, 148.62, 147.84, 147.15, 146.74, 146.65, 146.54, 146.50, 145.36, 143.06, 143.03, 142.79, 138.90, 138.51, 135.82, 129.18, 128.80, 125.38, 125.27, 125.15, 121.32, 120.91, 120.86, 117.12, 117.09, 116.77, 116.35, 116.18, 115.90, 115.22, 115.12, 114.56, 110.01, 33.66, 33.61, 32.97; HRMS (m/z, ESI+) Calcd for C39H31IrN9 818.2332 [M + H+], found: 818.2313.
Device fabrication and characterization.
PHOLEDs were grown on pre-cleaned glass substrates coated with 80-nm-thick indium tin oxide (ITO) by vacuum thermal evaporation in a chamber with a base pressure 6 × 10−7 torr. The devices consist of 10 nm MoO3 doped at 15 vol% in 9-(4-tert-butylphenyl)-3,6-bis(triphenylsilyl)-9H-carbazole (CzSi) as a hole injection layer (HIL)/5 nm CzSi hole-transport layer (HTL)/5 nm Ir(C∧C:)3-based electron and exciton blocking layer (EBL)/40 nm Ir(pmp)3 doped in TSPO1 to form the EML/5 nm TSPO1 hole and exciton blocking layer (HBL)/30 nm 1,3,5-tris(1-phenyl-1H-benzimidazol-2-yl)benzene (TPBi) electron-transport layer (ETL)/1.5 nm 8-hydroxyquinolinolato-Li electron injection layer (EIL)/100 nm Al (cathode). The devices were patterned using a shadow mask with an array of circular openings, resulting in contacts with a measured diameter of d = 430 μm. The standard deviation for a population of more than 20 devices leads to a variation in area of ∼2%. The EBLs used for the fac- and mer-Ir(pmp)3-based devices were fac-Ir(pmp)3 and fac-Ir(pmb)3, respectively (see Fig. 1a). The current density–voltage–luminance (J–V–L) characteristics were measured using a parameter analyser (HP4145, Hewlett–Packard) and a calibrated photodiode (FDS1010-CAL, Thorlab) according to standard procedures43. The emission spectra at J = 10 mA cm−2 were recorded using a calibrated spectrometer (USB4000, Ocean Optics) coupled to the device with an optical fibre.
Probing the recombination zone.
The exciton density, N(x), in the EML as a function of distance, x, from the EBL/EML interface was determined by measuring the relative emission intensity from a 1.5-nm-thick ‘sensing’ layer comprised of 5 vol% doped red-emitting phosphor—that is, iridium (III) bis (2-phenyl quinolyl-N, C2′) acetylacetonate (PQIr), inserted at different positions within the EML, as previously17.
EQE modelling.
Based on triplet–triplet and triplet–polaron annihilation dynamics, EQE versus J is modelled using:


where n is the electron density and N is the triplet density, γ = q(μn + μp)/ɛɛ0 is the Langevin recombination rate, and μn and μp are the respective electron and hole mobilities in the doped EML. ɛ = 3 is the dielectric constant, q is the charge, x is position, t is time, G is the charge generation rate, and kTT and kTP are the triplet–triplet and triplet–polaron annihilation rates, respectively. Here, G(x) = J/q ⋅ R(x), where R is the measured recombination profile in Fig. 4b, different from previous assumptions based on the constant recombination zone width16,18. Equation (1) assumes that p = n in the EML, and τ is obtained from the transient phosphorescence decay of Ir(pmp)3 doped into TSPO1. N(x) in equation (2) is obtained in steady state and integrated over the EML to obtain N. Then, N/J is normalized by the EQE at J = 0.1 mA cm−2 and fitted to J = 100 mA cm−2 (Fig. 5d, lines), where bimolecular quenching is active.
References
Baldo, M. A. et al. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 395, 151–154 (1998).
Baldo, M. A., Lamansky, S., Burrows, P. E., Thompson, M. E. & Forrest, S. R. Very high-efficiency green organic light-emitting devices based on electrophosphorescence. Appl. Phys. Lett. 75, 4–6 (1999).
Giebink, N. C. et al. Intrinsic luminance loss in phosphorescent small-molecule organic light emitting devices due to bimolecular annihilation reactions. J. Appl. Phys. 103, 044509 (2008).
Sajoto, T. et al. Blue and near-UV phosphorescence from iridium complexes with cyclometalated pyrazolyl or N-heterocyclic carbene ligands. Inorg. Chem. 44, 7992–8003 (2005).
Schildknecht, C. et al. Novel deep-blue emitting phosphorescent emitter. Proc. SPIE Int. Soc. Opt. Eng. 5937, 59370E (2005).
Hopkinson, M. N., Richter, C., Schedler, M. & Glorius, F. An overview of N-heterocyclic carbenes. Nature 510, 485–496 (2014).
Chang, C.-F. et al. Highly efficient blue-emitting iridium(III) carbene complexes and phosphorescent OLEDs. Angew. Chem. Int. Ed. 47, 4542–4545 (2008).
Chiu, Y.-C. et al. En route to high external quantum efficiency (∼12%), organic true-blue-light-emitting diodes employing novel design of iridium(III) phosphors. Adv. Mater. 21, 2221–2225 (2009).
Sasabe, H. et al. High-efficiency blue and white organic light-emitting devices incorporating a blue iridium carbene complex. Adv. Mater. 22, 5003–5007 (2010).
Hsieh, C.-H. et al. Design and synthesis of iridium bis(carbene) complexes for efficient blue electrophosphorescence. Chem. Eur. J. 17, 9180–9187 (2011).
Lu, K.-Y. et al. Wide-Range color tuning of iridium biscarbene complexes from blue to red by different N∧N ligands: An alternative route for adjusting the emission colors. Adv. Mater. 23, 4933–4937 (2011).
Fleetham, T., Wang, Z. & Li, J. Efficient deep blue electrophosphorescent devices based on platinum(II) bis(n-methyl-imidazolyl)benzene chloride. Org. Electron. 13, 1430–1435 (2012).
Lee, S. et al. Deep-blue phosphorescence from perfluoro carbonyl-substituted iridium complexes. J. Am. Chem. Soc. 135, 14321–14328 (2013).
Hang, X.-C., Fleetham, T., Turner, E., Brooks, J. & Li, J. Highly efficient blue-emitting cyclometalated platinum(II) complexes by judicious molecular design. Angew. Chem. Int. Ed. 52, 6753–6756 (2013).
Fleetham, T., Li, G., Wen, L. & Li, J. Efficient ‘Pure’ blue OLEDs employing tetradentate Pt complexes with a narrow spectral bandwidth. Adv. Mater. 26, 7116–7121 (2014).
Baldo, M. A., Adachi, C. & Forrest, S. R. Transient analysis of organic electrophosphorescence. II. Transient analysis of triplet–triplet annihilation. Phys. Rev. B 62, 10967–10977 (2000).
Zhang, Y., Lee, J. & Forrest, S. R. Tenfold increase in the lifetime of blue phosphorescent organic light-emitting diodes. Nature Commun. 5, 5008 (2014).
Erickson, N. C. & Holmes, R. J. Engineering efficiency roll-off in organic light-emitting devices. Adv. Funct. Mater. 24, 6074–6080 (2014).
Park, M. S. & Lee, J. Y. Indolo acridine-based hole-transport materials for phosphorescent OLEDs with over 20% external quantum efficiency in deep blue and green. Chem. Mater. 23, 4338–4343 (2011).
Holmes, R. J. et al. Saturated deep blue organic electrophosphorescence using a fluorine-free emitter. Appl. Phys. Lett. 87, 243507 (2005).
Deaton, J. C., Young, R. H., Lenhard, J. R., Rajeswaran, M. & Huo, S. Photophysical properties of the series fac- and mer-(1-Phenylisoquinolinato-N∧C2′)x(2-phenylpyridinato-N∧C2′)3−x iridium(III) (x = 1–3). Inorg. Chem. 49, 9151–9161 (2010).
Tamayo, A. B. et al. Synthesis and characterization of facial and meridional tris-cyclometalated iridium(III) complexes. J. Am. Chem. Soc. 125, 7377–7387 (2003).
Sajoto, T. et al. Temperature dependence of blue phosphorescent cyclometalated Ir(III) complexes. J. Am. Chem. Soc. 131, 9813–9822 (2009).
Lauerhaas, J. M. et al. Reversible luminescence quenching of porous silicon by solvents. J. Am. Chem. Soc. 114, 1911–1912 (1992).
Xiao, L. et al. Recent progresses on materials for electrophosphorescent organic light-emitting devices. Adv. Mater. 23, 926–952 (2011).
Yook, K. S. & Lee, J. Y. Organic materials for deep blue phosphorescent organic light-emitting diodes. Adv. Mater. 24, 3169–3190 (2012).
Djurovich, P. I., Mayo, E. I., Forrest, S. R. & Thompson, M. E. Measurement of the lowest unoccupied molecular orbital energies of molecular organic semiconductors. Org. Electron. 10, 515–520 (2009).
Jeon, S. O., Jang, S. E., Son, H. S. & Lee, J. Y. External quantum efficiency above 20% in deep blue phosphorescent organic light-emitting diodes. Adv. Mater. 23, 1436–1441 (2011).
Reineke, S., Walzer, K. & Leo, K. Triplet-exciton quenching in organic phosphorescent light-emitting diodes with Ir-based emitters. Phys. Rev. B 75, 125328 (2007).
Su, S.-J., Gonmori, E., Sasabe, H. & Kido, J. Highly efficient organic blue-and white-light-emitting devices having a carrier- and exciton-confining structure for reduced efficiency roll-off. Adv. Mater. 20, 4189–4194 (2008).
Seino, Y., Sasabe, H., Pu, Y.-J. & Kido, J. High-performance blue phosphorescent OLEDs using energy transfer from exciplex. Adv. Mater. 26, 1612–1616 (2014).
Zhang, Q. et al. Efficient blue organic light-emitting diodes employing thermally activated delayed fluorescence. Nature Photon. 8, 326–332 (2014).
Yang, C.-H. et al. Deep-blue-emitting heteroleptic iridium(III) complexes suited for highly efficient phosphorescent OLEDs. Chem. Mater. 24, 3684–3695 (2012).
You, Y. & Nam, W. Photofunctional triplet excited states of cyclometalated Ir(III) complexes: Beyond electroluminescence. Chem. Soc. Rev. 41, 7061–7084 (2012).
Tsuchiya, K., Ito, E., Yagai, S., Kitamura, A. & Karatsu, T. Chirality in the photochemical mer → fac geometrical isomerization of Tris(1-phenylpyrazolato,N,C2′)iridium(III). Eur. J. Inorg. Chem. 2009, 2104–2109 (2009).
Kober, E. M., Sullivan, B. P. & Meyer, T. J. Solvent dependence of metal-to-ligand charge-transfer transitions. Evidence for initial electron localization in MLCT excited states of 2,2′-bipyridine complexes of ruthenium(II) and osmium(II). Inorg. Chem. 23, 2098–2104 (1984).
Reineke, S., Rosenow, T. C., Lüssem, B. & Leo, K. Improved high-brightness efficiency of phosphorescent organic LEDs comprising emitter molecules with small permanent dipole moments. Adv. Mater. 22, 3189–3193 (2010).
Padmaperuma, A. B., Sapochak, L. S. & Burrows, P. E. New charge transporting host material for short wavelength organic electrophosphorescence: 2,7-Bis(diphenylphosphine oxide)-9,9-dimethylfluorene. Chem. Mater. 18, 2389–2396 (2006).
Calcagno, P. et al. Understanding the structural properties of a homologous series of bis-diphenylphosphine oxides. Chem. Eur. J. 6, 2338–2349 (2000).
Kawamura, Y. et al. 100% phosphorescence quantum efficiency of Ir(III) complexes in organic semiconductor films. Appl. Phys. Lett. 86, 071104 (2005).
Erk, P. et al. 11.2: Efficient deep blue triplet emitters for OLEDs. SID Symp. Dig. Tech. Pap. 37, 131–134 (2006).
Shi, F., Xu, X., Zheng, L., Dang, Q. & Bai, X. Method development for a pyridobenzodiazepine library with multiple diversification points. J. Comb. Chem. 10, 158–161 (2008).
Forrest, S. R., Bradley, D. D. C. & Thompson, M. E. Measuring the efficiency of organic light-emitting devices. Adv. Mater. 15, 1043–1048 (2003).
Acknowledgements
This work was supported by the Air Force Office of Scientific Research (AFOSR) and Universal Display Corporation.
Author information
Authors and Affiliations
Contributions
J.L. designed, fabricated and optimized the PHOLEDs, and analysed the optical and electrical properties of materials with S.R.F., M.E.T. and P.I.D. H.-F.C., T.B. and M.E.T. synthesized and measured the photophysical and electrochemical properties of materials. C.C. provided EQE roll-off theory and modelling. S.R.F. supervised the project, analysed data, and wrote the manuscript with J.L.
Corresponding author
Ethics declarations
Competing interests
M.E.T. and S.R.F. have an equity interest in one of the sponsors of this work (Universal Display Corp.).
Supplementary information
Supplementary Information
Supplementary Information (PDF 2688 kb)
Rights and permissions
About this article

Cite this article
Lee, J., Chen, HF., Batagoda, T. et al. Deep blue phosphorescent organic light-emitting diodes with very high brightness and efficiency. Nature Mater 15, 92–98 (2016). https://doi.org/10.1038/nmat4446
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmat4446
This article is cited by
-
High-efficiency and stable short-delayed fluorescence emitters with hybrid long- and short-range charge-transfer excitations
Nature Communications (2023)
-
Double boron–oxygen-fused polycyclic aromatic hydrocarbons: skeletal editing and applications as organic optoelectronic materials
Nature Communications (2023)
-
Electroluminescence and hyperphosphorescence from stable blue Ir(III) carbene complexes with suppressed efficiency roll-off
Nature Communications (2023)
-
Efficient room-temperature phosphorescence of covalent organic frameworks through covalent halogen doping
Nature Chemistry (2023)
-
Visualizing electroluminescence process in light-emitting electrochemical cells
Nature Communications (2023)
