
Abstract
HER2 amplification-associated molecular alterations and clinicopathologic features in colorectal cancers (CRCs) have not been well established. In this study, we assessed the prevalence of HER2 amplification and microsatellite instability (MSI) status of 992 patients with primary CRC. In addition, molecular alterations of HER2 amplified and unamplified CRCs were examined and compared by next-generation sequencing. HER2 amplifications were found in 41 (4.1%) of 992 primary CRCs. HER2 amplification was identified in 1.0% of the right colonic tumors, 5.1% of the left colonic tumors, and 4.8% of the rectal tumors. Approximately 95% of HER2 amplification was observed in the left colon and rectum. Seven (87.5%) of eight metastatic tumors showed HER2 amplification. Most clinicopathologic features were unrelated to HER2 amplification except tumor size and MSI status. All 41 HER2 amplified CRCs were microsatellite stable. In a molecular analysis of frequently identified somatic mutations in CRCs, HER2 amplified CRCs showed a lower rate of KRAS mutations (24.4%) but a higher rate of TP53 mutations (83%) than unamplified CRCs. No BRAF and NRAS mutations were identified in HER2 amplified CRCs. Our study suggests that HER2 amplified CRCs are mutually exclusive of MSI and harbor less frequent KRAS/NRAS/BRAF mutations but frequent T53 mutations.
Similar content being viewed by others


Introduction
The frequency of HER2 overexpression/amplification ranges from 1.3 to 6.3% of unselected colorectal cancers (CRCs)1,2,3. HER2 amplification leading to HER2 protein overexpression is associated with downstream activated signaling pathways, leading to dysregulated cell proliferation, including breast cancer, esophagogastric cancer, and CRC4. Recently, the U.S. Food and Drug Administration approved tucatinib + trastuzumab combination therapy for patients with HER2 amplified CRC5. Although the prevalence of HER2 amplification in CRCs is relatively lower than in other cancers, detecting HER2 amplification in CRCs becomes inevitable for selecting patients who may benefit from HER2-targeted therapy.
Microsatellite instability (MSI) is a hypermutable phenotype caused by mismatch repair deficiency, which accounts for 10–15% of all CRCs6. The MSI phenotype involves the primary loss of function of proteins that usually repair DNA base-pair mismatches, mainly MLH1, MSH2, MSH6, and PMS2. In association with other molecular alterations identified in CRCs, the microsatellite instability-high (MSI-H) phenotype has shown to be significantly associated with BRAF V600E mutations but tends to correlate inversely with KRAS mutation7. In contrast, the possible correlation between HER2 amplification and MSI status has not been well investigated. A few molecular studies of CRCs and gastric cancers suggest that most HER2 amplified tumors were microsatellite stable (MSS), representing a negative correlation between HER2 amplification and MSI in gastrointestinal cancers8,9.
According to the National Comprehensive Cancer Network (NCCN) guideline, all patients with metastatic CRC who are eligible for anti-epidermal growth factor receptor (EGFR) inhibitors should have the tumor tissue tested for BRAF and RAS mutations10. Because BRAF and RAS mutations, including KRAS and NRAS, are associated with primary resistance to anti-EGFR therapy. The current guideline recommends that only patients with RAS wild type should be treated with anti-EGFR inhibitors after evaluation of RAS mutations. Therefore, evaluation of RAS mutations in HER2 amplified CRCs can provide further information to select therapeutic options, including HER2-targeted therapy.
The correlations between HER2 status and different clinicopathologic features have been controversial. Some studies suggest that HER2 overexpression is associated with aggressive biological behavior, which includes profound invasion, lymphatic invasion, lymph node metastasis, distant metastasis, and distal colon location3,11,12. In contrast, other studies demonstrated no correlations between clinicopathologic features, including tumor location and HER2 overexpression/amplification13,14,15. Thus, we investigate to find any correlations between HER2 amplification and clinicopathologic features in a cohort study of 992 patients with primary CRCs.
The main purpose of this study was to better elucidate the prevalence of HER2 expression and amplification and associated clinicopathologic features in patients with primary unselected CRC. After the selection of HER2 amplified CRC cases, microsatellite instability status and molecular alterations of HER2 amplified and unamplified CRCs after matching tumor sites were compared by next-generation sequencing.
Results
Expression and amplification of HER2
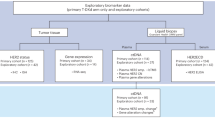
A flow chart of HER2 expression and amplification of 992 primary CRCs revealed by immunohistochemistry and dual-colored in situ hybridization (DISH) is illustrated in Fig. 1. Among 992 primary CRCs, there were 149 (15%) tumors with 1 + labeling, 50 (5.0%) tumors with 2 + labeling, and 33 (3.3%) tumors with 3 + labeling. DISH was performed on 232 tumors with 1 + , 2 + , and 3 + labeling. All 33 (3.3%) tumors with 3 + labeling revealed high HER2 amplifications on DISH. Among 50 (5%) tumors with 2 + labeling, 8 (0.8%) tumors were found to have HER2 amplifications on DISH. Among 41 (4.1%) HER2 amplified tumors, there were 36 (3.6%) tumors with high amplifications and 5 (0.5%) tumors with low amplifications on DISH. During follow-up, eight patients underwent surgical resections of metastatic lesions. Additional immunohistochemistry and DISH were performed on eight metastatic tumors in the liver and lung. Seven (87.5%) of eight metastatic tumors showed HER2 amplification on DISH. The concordance rate of HER2 amplification between the primary and metastatic CRCs was 87.5%. Immunohistochemistry and DISH for HER2 on primary and metastatic CRC tumors are described in Fig. 2.

A flow chart showing each step of detecting HER2 expressed and amplified tumors in 992 invasive CRCs using immunohistochemistry and DISH. Among 992 unselected CRCs, there were 149 (15%) tumors with 1 + labeling, 50 (5%) tumors with 2 + labeling, and 33 (3.3%) tumors with 3 + labeling by immunohistochemistry. HER2 DISH was performed on tumors with all 1 + , 2 + , and 3 + labeling. Finally, 41(4.1%) tumors were found to be HER2 amplified.

Representative micrographs of HER2 immunohistochemistry and DISH on the primary and metastatic CRCs are shown. (A) The primary CRC reveals a diffuse and strong staining pattern (3 +) of HER2 immunostaining at low magnification (4×). (B) The corresponding primary tumor shows diffusely located individuals and clusters of black dots within neoplastic cells on DISH. (C) At high magnification (40×), neoplastic cells exhibit intense membranous staining (3 +) of HER2 (left) and highly clustered black dots (right) representing amplified HER2 gene signals. (D) Seven of eight metastatic CRCs harbor HER2 amplification on DISH, representing 87.5% of the concordance rate between the primary and metastatic CRCs. The metastatic CRC in the liver displays a diffuse and strong staining pattern (3 +) of HER2 at low magnification (4×). (E) The corresponding metastatic tumor shows diffuse and strong black dots on DISH. (F) At high magnification (20×), neoplastic cells within the liver parenchyma reveal a strong membranous staining pattern (3 +) of HER2 (left) and highly clustered black dots (right) representing amplified HER2 gene signals.
Clinicopathologic features and MSI status of Her2 amplified and unamplified CRCs
Among 992 patients with primary CRCs, forty-one (4.1%) patients had HER2 amplified CRC. HER2 amplifications were identified in 1.0% (2/193) of the right colonic tumors, 5.1% (20/394) of the left colonic tumors, and 4.8% (19/400) of the rectal tumors. In clinicopathologic features of 41 patients with HER2 amplified CRC, the patients consisted of twenty-four (58.5%) men and seventeen (41.5%) women (male to female ratio: 1.4), with a mean age of 60 years. One patient had a history of ulcerative colitis. Four patients had a history of prior sigmoid colon cancer, lung adenocarcinoma, early gastric adenocarcinoma, and gastric gastrointestinal stromal tumor. Six (14.6%) patients with HER2 amplified CRC received neoadjuvant chemoradiation before the surgery. The median tumor size was 3.5 cm (0.8–7.8 cm in greatest dimension). There was one case with two synchronous tumors in the cecum and ascending colon/right-sided colon. The most common primary tumor site was the left colon (48.8%), followed by the rectum (46.3%) and the right colon (4.9%). In twenty (48.8%) amplified left-sided tumors, seventeen (85%) were observed in the sigmoid colon. In nineteen (46.3%) amplified rectal tumors, eleven (57.9%) were observed in the upper rectum, a median of 11 cm (range, 7–15 cm) located above the anal verge; eight (42.1%) tumors were found in the lower rectum, a median of 3.2 cm (range, 2–5.9 cm) above the anal verge. Two (4.9%) HER2 amplified tumors were observed in the ascending colon. The anastomotic sites of HER2 amplified primary CRCs are illustrated in Fig. 3. Twenty-five (60.1%) HER2 amplified tumors were in the T3 and T4 categories. Metastatic disease was noted in 4 (9.8%) patients at the time of diagnosis, with the liver being the most common organ involved. Fourteen (34.1%) patients with HER2 amplified CRC were diagnosed with pathologic stages III and IV. In the comparison analysis of HER2 amplified and unamplified CRCs, there were no statistical differences in most clinicopathologic features between the two tumor groups except tumor size and MSI status. All 41 HER2 amplified CRCs were MSS phenotypes revealed by MSI testing compared to 100 (10.5%) MSI-H tumors in 951 unamplified CRCs. A comparison analysis of the clinicopathologic features of HER2 amplified and unamplified tumor groups is detailed in Table 1.

This illustration shows the anastomotic site where HER2 amplified tumors occur in the colorectum. The most common primary tumor site was the left colon (48.8%), followed by the rectum (46.3%) and the right colon (4.9%). In HER2 amplified left colonic tumors, seventeen (85%) were observed in the sigmoid colon. Moreover, in HER2 amplified rectal tumors, eleven (57.9%) occurred in the upper rectum, a median of 11 cm (range, 7–15 cm) located above the anal verge. All two (4.9%) HER2 amplified tumors were found in the ascending colon.
Molecular alterations between HER2 amplified and unamplified CRCs
All forty-one primary CRCs with HER2 amplification on DISH were examined using the 88-gene panel. Copy numbers of HER2 ranged from 5 to 43 with a mean copy number of 13. There was no discordance between the results of DISH and next-generation sequencing in HER2 amplification (100% concordance rate). No activating BRAF mutations were detected. Activating KRAS mutations were identified in 24.4% (10/41) amplified tumors, predominantly in codons 12 and 13, including p.G12D, p.G12V, and p.G13D. Other RAS genes, such as NRAS and HRAS mutations, were not detected. TP53 mutations were identified in 82.9% (34/41) of amplified tumors without consistently mutated hot spots. APC mutations were found in 51.2% (21/41) of amplified tumors without mutated hot spots. PIK3CA, ERBB2/3, SMAD4, PTEN, and FBXW7 mutations were detected in 17.1% (7/41), 7.3% (3/41), 4.9% (2/41), 2.4% (1/41), and 7.3% (3/41) of amplified tumors. Additionally, mutations in various genes, including CDK4, ATM, KIT, and so on, were occasionally detected. The majority of these mutations are of unknown functional consequences. Somatic mutations and gene alterations identified in 41 HER2 amplified CRCs are listed in supplementary Table 1.
Due to the different prevalences of somatic mutations depending on the anatomical site, molecular data for two hundred CRCs matched by tumor sites were retrospectively collected and analyzed for comparison. A detailed flow chart outlining the selection of 200 CRCs as a control group after matching tumor sites with 42 HER2 amplified CRCs is illustrated in Fig. 4. Compared with the 200 unamplified CRCs, TP53 mutations were more commonly observed in HER2 amplified tumors than unamplified tumors (83% vs 58%; p = 0.0002). However, KRAS mutations were less frequently identified in the HER2 amplified tumors than in unamplified tumors (24% vs 49%; p = 0.0004). BRAF and NRAS mutations were not detected in HER2 amplified tumors but were observed in 4% and 6% unamplified tumors. There were no statistical differences in the frequencies of other genes, including APC, PIK3CA, SMAD4, ERBB2/3, PTEN, and FBXW7, which have been reported mutated in CRCs by next-generation sequencing. The somatic mutations and copy numbers of genes identified in the 41 HER2 amplified CRCs are detailed in Fig. 5A. The results of the comparative analysis of frequently identified mutations in HER2 amplified and unamplified CRCs after matching tumor sites are depicted in Fig. 5B.

In order to compare molecular differences between HER2 amplified and unamplified CRCs, previous molecular data for 314 HER2 unamplified CRCs were available. As there are known differences in the frequencies and distributions of mutations in key oncogenes and tumor suppressors between right and left-sided CRCs, a control group of HER2 unamplified CRCs was selected after matching tumor sites with 41 HER2 amplified CRCs. The most common HER2 amplified tumor site was the left colon (48.8%), followed by the rectum (46.3%) and the right colon (4.9%). A flow chart outlines the selection process of 200 CRCs as a control group after matching tumor sites. The 200 CRCs selected consisted of 5% right-sided colon cancer, 50% left-sided colon cancer, and 45% rectal cancers, with varying frequencies of KRAS/NRAS/BRAF mutations.

(A) Somatic mutational profile and copy number variations of HER2 in HER2 amplified CRCs are illustrated. The most frequently identified somatic mutations were found in the TP53 gene, followed by the APC gene. Predominantly, missense mutations were found in KRAS and TP53 mutations. In contrast, mostly truncating and inframe mutations were observed in APC mutations. Copy numbers of HER2 ranged from 5 to 43, with a mean copy number of 13. (B) Comparative analysis of frequently identified somatic mutations between HER2 amplified and unamplified CRCs. No BRAF and NRAS mutations were identified in HER2 amplified CRCs. HER2 amplified CRCs harbored KRAS mutations less frequently than unamplified CRCs (24% vs 59%; p < 0.0001). In contrast, TP53 mutations were more frequently identified in HER2 amplified CRC than unamplified ones (83% vs 62%; p = 0.0014). There were no differences in frequencies of other gene mutations. All HER2 amplified CRCs were MSS phenotypes.
Clinical outcome
Follow-up data were available for all 992 patients with primary CRC. The 1-, 3-, and 5-year overall survival (OS) rates for 992 patients were 95%, 83%, and 77%, respectively, with a median follow-up of 52 months (range 1–60 months). Kaplan–Meier survival analysis demonstrated a poor tumor differentiation, high T category, lymphovascular invasion, high N category, distant metastasis at diagnosis (M category), and advanced pathologic TNM stage (all, p < 0.0001) to be significantly associated with shorter OS. Regarding potential HER2 amplification and MSI-H status related to survival, there were no significant differences in OS rates between groups with HER2 amplified and unamplified tumors (p = 0.827) or groups with MSS/MSI-L and MSI-H phenotypes (p = 0.065). Kaplan–Meier survival analysis for the T category, pathologic TNM stage, HER2 amplification, and MSI status are illustrated in Fig. 6.

Kaplan–Meier cumulative survival curves. (A) Survival according to T category (p < 0.001); (B) Survival according to pathologic TNM stage (p < 0.0001); (C) Survival according to HER2 amplified and unamplified tumors (p = 0.827); (D) Survival according to MSI-H and MSS/MSI-L tumors (p = 0.065).
In the survival analysis of 41 patients with HER2 amplified CRC, lymphovascular invasion (p = 0.015), lymph node metastasis (p = 0.018), and pathologic AJCC stage III/IV (p = 0.014) were statistically associated with shorter OS. Regarding potential molecular alterations related to survival, there were no differences in OS rates between groups with mutated and wild type TP53, KRAS, APC, PIK3CA, and FBXW7.
Discussion
The frequency of HER2 overexpression/amplification has been reported in 1.3–6.3% of unselected CRCs1,2,3. The Cancer Genome Atlas (TCGA) project detected HER2 alterations in approximately 7% of patients with CRC, especially in RAS and BRAF wild type tumors16. In this series of 992 patients with primary CRC, forty-one (4.1%) patients were found to have HER2 amplification. HER2 amplifications were identified in 1.0% (2/193) of the right colonic tumors, 5.1% (20/394) of the left colonic tumors, and 4.8% (19/400) of the rectal tumors. A retrospective molecular analysis of 1730 CRCs demonstrated that CRC tumors with HER2 amplification were preferentially located on the left side of the colon17. Our study also found that the most common site showing HER2 amplification was the left colon (48.8%), followed by the rectum (46.3%). Previous and current studies support that HER2 amplification differs depending on the location of the primary tumor, which tends to be located more frequently in the left colon and rectum. Based on our findings, it would be beneficial to do immunohistochemistry for HER2 as a screening test on the left-sided CRCs where 95.1% of HER2 amplifications occur.
There have been conflicting studies on the relationship between HER2 status and clinicopathologic features in patients with CRC3,11,13,14. However, our study demonstrated that there were no significant differences in most clinicopathologic features between amplified and unamplified tumor groups, except for tumor size and MSI status. Moreover, the prognostic significance of HER2 status in CRCs is disputable. Some studies suggest that HER2 positivity is related to poor OS, while others have not found any correlation between HER2 positivity and prognosis15,18,19,20. The present study showed no statistical differences in OS between groups of HER2 amplified and unamplified CRCs. Although further studies are needed to evaluate the prognostic effect of HER2 status in CRCs, adding anti-HER2 inhibitor alone or combination therapy is a promising treatment option for better clinical outcomes in patients with advanced HER2 amplified CRC.
The rate of MSI-H status in primary CRCs is reported to be 10–15%6. The rate of MSI-H phenotype in this study was 10.1% of 992 primary CRCs, which number is within the reported ranges. The relationship between HER2 amplification and MSI status has yet to be well elucidated. In the MyPathway study, 28 patients with HER2 amplified metastatic CRCs were MSS21. Another molecular study demonstrated that none of the 16 HER2 amplified CRCs were MSI-H22. In this series, all 41 (4.1%) HER2 amplified CRCs were MSS phenotypes, and all 100 (10.2%) of MSI-H CRCs were unamplified. The prior and current studies suggest that the relationship between HER2 amplification and MSI-H phenotype in metastatic and primary CRCs is mutually exclusive.
BRAF mutation is reported in approximately 5–10% of CRCs23. Patients with BRAF mutated CRC tend to respond poorly to chemotherapy and progress rapidly with a poorer prognosis compared to those with BRAF wild-type CRC6,24. Additionally, these patients develop resistance to anti-EGFR inhibitors due to activation of the MAPK pathway downstream of EGFR25. Therefore, BRAF mutations are considered to be negative predictors of anti-EGFR therapy26. Previous studies have also found that 91% of BRAF mutations are present in MSI-H CRCs with hMLH1 methylation27,28. In this series, 4% of BRAF mutations were found in 200 unamplified CRCs, and 90% of BRAF mutated tumors were MSI-H. In contrast, no BRAF mutations were identified in all 41 HER2 amplified CRCs, indicating a negative relationship between HER2 amplification and BRAF mutation in CRCs.
The prevalence of KRAS mutations is reported to be 30–50% in CRC29. KRAS mutations are predominantly clustered in exon 2, in codons 12 (near 80% of all KRAS mutations), and are less frequent in exons 3 (codons 59 and 61) and 4 (codons 117 and 146) 30,31,32. NRAS mutations are uncommon, accounting for 3–5% of CRCs, predominantly in exons 2, 3, and 433. So far, KRAS and NRAS mutations are associated with clinical resistance to anti-EGFR therapy, which is not recommended for patients with these mutations10. However, the incidence of RAS mutations in HER2 amplified CRCs has not been well described. In a study of HER2 amplification and KRAS/BRAF mutation in stage II-III CRC patients, HER2 amplifications were less frequent in tumors with KRAS/BRAF mutations (observed in 0.2–1.4% of tumors harboring KRAS/BRAF mutations)1. Consistent with this, our study demonstrated that HER2 amplified CRCs harbored less frequent KRAS mutations than unamplified CRCs (24.4% vs 49%; p = 0.0004).
HER2 amplification in CRCs is significantly associated with resistance to anti-EGFR inhibitors and also related to inferior survival outcomes in patients with HER2 positive tumor exposed to chemotherapy with anti-EGFR inhibitors34,35. Therefore, in addition to RAS/RAF molecular tests, HER2 screening is essential for selecting efficacious treatment options for patients with metastatic CRC. According to the updated NCCN guideline, patients with HER2 amplified and RAS/RAF wild type CRC are eligible candidates for fam-trastuzumab deruxtecan-nxki or a combination of trastuzumab with pertuzumab or lapatinib or tucatinib35. Our study demonstrated that approximately 75.6% of patients with HER2 amplified CRC had RAS/RAF wild type, suggesting that they may benefit from a combination therapy of anti-HER2 inhibitors in advanced stages of the disease.
Approximately 60% of CRCs are reported to harbor TP53 mutation36. However, the prevalence rate of TP53 mutation in CRC varies depending on the anatomical site and tumor molecular subtype37. The frequency of TP53 mutations is higher in distal colon and rectal tumors, while proximal colon tumors with MSI-H and methylator phenotypes have lower frequencies of TP53 mutations37,38.
Frequent T53 mutations in HER2 amplified CRCs, up to 83%, were found in this series. In breast cancers, the frequency of TP53 mutations was reported to be more common in HER2 positive than HER2 negative tumors39,40. However, in this series of HER2 amplified CRCs, it is unclear whether frequent TP53 mutations are associated with HER2 amplification or are due to a predominant anastomotic site/distal colorectum of HER2 amplified tumors. Moreover, it is well-known that TP53 mutations have been associated with a poorer prognosis in CRCs36,41. However, we could not find any significant differences in survival between TP53 mutated versus wild type patients with HER2 amplified CRC, possibly due to a small number of cases and limited follow-up period. Therefore, further validation studies, including a large-scale series, are needed to investigate TP53 mutations and associated clinical outcomes in HER2 amplified CRCs.
The current study has a few limitations. It is a retrospective study conducted at a single institution. The differences in clinicopathologic features and rates of HER2 amplification may originate from selection biases and interpretation of HER2 amplification by different methodologies, fluorescence in situ hybridization versus. DISH. However, the concordance rate of HER2 amplification between results of FISH and next-generation sequencing was 100% in this study. In addition, molecular data for a control group was retrospectively collected from patients who underwent next-generation sequencing with different numbers of gene panels (80–559 gene panels). Therefore, only frequently identified somatic mutations in CRCs between the study and control groups were compared.
In summary, HER2 amplifications were found in 41 (4.1%) of 992 primary CRCs. All 41 HER2 amplified tumors were MSS. In addition, HER2 amplified CRCs showed a lower rate of KRAS mutations (24.4%) but a higher rate of TP53 mutations (83%) than unamplified CRCs. No BRAF and NRAS mutations were identified. Our study suggests that HER2 amplified CRCs are mutually exclusive of microsatellite instability and harbor less frequent KRAS/NRAS/BRAF mutations but frequent TP53 mutations.
Methods
Patient selection and clinicopathologic evaluation
We reviewed pathology records of patients diagnosed with primary colorectal adenocarcinoma from the Pathology Department at JNUH. Thousand twelve surgically resected cases were initially searched and reviewed from 2017 to 2022. Among them, cases with colorectal adenocarcinoma in situ (Tis) were excluded. A total of nine hundred ninety-two surgical cases of invasive CRCs were selected. Histologic findings were re-evaluated, including tumor size, tumor differentiation, depth of invasion, lymphovascular invasion, perineural invasion, regional lymph node metastasis, distant metastasis, and pathologic TNM staging. Tumors were restaged according to the 8th edition of the American Joint Committee on Cancer (AJCC) TNM staging system for CRCs after the clinicopathologic features and radiologic findings had been re-reviewed42. Demographic and clinical information, including sex, age, type of surgery, tumor site, date of diagnosis, preoperative neoadjuvant therapy, and last follow-up status, were collected from reviews of the patient’s medical records. This study was approved by the institutional review board (IRB) of Jeju National University Hospital (IRB number: JEJUNUH 2022-01-013), with written patient consent deemed unnecessary.
Immunohistochemistry and DISH for HER2
Immunohistochemistry was performed on a representative tissue section of each case using the avidin–biotin method. The primary antibody was against HER2/neu (rabbit monoclonal antibody clone 4B5, prediluted, Ventana Roche, USA). An automated stainer (Ventana Medical Systems, AZ, USA) was used per the manufacturer’s protocol. The level of HER2 expression was evaluated according to Hofmann’s scoring system used for surgical specimens as follows: 0, no reactivity or membranous activity in < 10% of tumor cells; 1 + , faint or barely detectable membranous reactivity in ≥ 10% of tumor cells; 2 + , weak to moderate complete, basolateral or lateral membranous reactivity in ≥ 10% of tumor cells; and 3 + , moderate to strong complete or basolateral membranous reactivity in ≥ 10% of tumor cells43.
After reviewing the immunohistochemical results of HER2 proteins, DISH was performed with cases with IHC scores of 1 + , 2 + , and 3 + . DISH was performed on a Bench Mark XT (Ventana Medical Systems) using the FDA-approved INFORM HER2 DISH DNA Probe Cocktail Assay (Ventana Medical Systems), which contains a digoxigenin (DIG)-labeled probe for the ERBB2 locus and a dinitrophenyl (DNP)-labeled probe for the centromeric region of chromosome 17. Briefly, 4-μm sections from each case were cut and deparaffinized. They were incubated in Cell Conditioning 2 (CC2) solution followed by protease digestion with ISH Protease 3. After applying 10 μl of INFORM HER2 Dual ISH DNA probe, the genomic DNA in tissue sections and the nick-translated HER2 probes were codenatured by heat treatment and hybridization. HER2 signals were detected using an UltraView DISH DNP and Red ISH DIG Detection Kit. The HER2/CEP17 ratio was determined by counting the HER2 and CEP17 signals in 20 nuclei for each section. HER2 gene amplification was defined as a DISH result showing a HER2/CEP17 ratio of ≥ 2.044. When the HER2/CEP17 ratio was between 1.8 and 2.2, twenty additional cells were recounted in an alternative area and added.
Microsatellite instability testing
Microsatellite instability (MSI) analysis was performed on all colorectal tumors by multiplex polymerase chain reaction (PCR) with five quasi-monomorphic mononucleotide repeat markers: BAT25, BAT26, D5S346, D17S250, and D2S123. Genomic DNA was isolated from paraffin-embedded tumor tissues using a QIAamp DNA Mini Kit (Qiagen, CA, USA). Each primer was end-labeled with one of the following fluorescent markers: FAM, HEX, or NED. An ABI Prism 3130 Genetic Analyzer (Applied Biosystems, CA, USA) was used to analyze the products, and allelic sizes were estimated by Genemapper 4.1 (Applied Biosystems, CA, USA). Tumors with allelic size variation in two or more microsatellite markers were deemed microsatellite instability-high (MSI-H), whereas tumors with allelic variations in one of the microsatellites were classified as microsatellite instability-low (MSI-L). If there were no allelic size variations, all microsatellites were considered microsatellite-stable (MSS).
Targeted next-generation sequencing
Deep targeted DNA sequencing was performed on all 41 HER2 amplified tumor samples. After a review of matched hematoxylin and eosin-stained slides, formalin-fixed and paraffin-embedded (FFPE) tissue blocks containing adequate tumor cellularity (> 70%) were selected by S.L. Genomic DNA was extracted using QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA, USA), and quantified using a Qubit dsDNA HS Assay Kit with the Qubit 1.0 fluorometer (Life Technologies, Carlsbad, CA, USA). A NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) was used to measure the concentration, and an initial quality check was performed using electrophoresis on 1% agarose gel, loading 0.5 µl of DNA solution. Deep targeted DNA sequencing was performed using the NextSeq 500 system (Illumina, San Diego, CA, USA) with an 88-gene panel mainly focusing on gastrointestinal cancers. Briefly, the cancer-related genes evaluated included ABL1, AKT1, AKT3, ALK, APC, BRAF, BRCA1, BRCA2, CDH1, CDKN2A, EGFR, ERBB2, ERBB3, FBXW7, GNAS, HRAS, IDH1, IDH2, KRAS, MLH1, NOTCH1, NRAS, PDGFRA, PIK3CA, PTEN, RB1, SMAD4, SMARCB1, TP53, VHL, WT1 among others. Using the hybrid capture method, DNA libraries were prepared using an Agilent SureSelectXT Target Enrichment Kit (Agilent Technologies, Santa Clara, CA, USA). DNA libraries that passed quality checks were sequenced using NextSeq 500. Library preparation and deep targeted sequencing were performed at Macrogen Incorporate (Seoul, Republic of Korea). Library preparation and targeted sequenced reads were mapped to the human reference genome hg38 using the Burrows-Wheeler Aligner-MEM algorithm with default options45. Variants were called by the Genome Analysis Tool Kit (GATK) v4.0.9.146 and annotated using SnpEff & SnpSift v4.3i47 and dbNSFP v2.9.348. False-positive variants were manually curated using Integrative Genomic Viewer49. Amplification of HER2 was defined as ≥ 5 copies of HER2 above the average ploidy of the tumor sample50. Among 951 unamplified CRCs, we retrospectively identified and collected results of next-generation sequencings with different gene panels (80–559 gene panels) of 200 primary CRCs after matching tumor sites as a control group to compare frequently identified somatic mutations with HER2 amplified CRCs.
Statistical analysis
Pearson’s chi-square test or Fisher’s exact test was applied to evaluate correlations between clinicopathologic variables and frequently mutated genes. Continuous variables were analysed using Student’s t-test or the Mann–Whitney U-test. Cumulative survival rates were calculated by the Kaplan–Meier method. Significant differences in survival status were evaluated using log-rank and Wilcoxon tests. The statistical analyses were performed using SPSS 24.0 software (SPSS Inc., Chicago, IL). A p-value < 0.05 was considered statistically significant.
Ethical approval and consent to participate
The study was conducted in accordance with the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Jeju National University Hospital (IRB File No. 2022-01-013). The informed written consent was also obtained from the subjects involved in this study.
Data availability
Data is provided within the manuscript and supplementary information files.
References
Richman, S. D. et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: Analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J. Pathol. 238, 562–570. https://doi.org/10.1002/path.4679 (2016).
Marx, A. H. et al. Heterogenous high-level HER-2 amplification in a small subset of colorectal cancers. Hum. Pathol. 41, 1577–1585. https://doi.org/10.1016/j.humpath.2010.02.018 (2010).
Seo, A. N. et al. HER2 status in colorectal cancer: Its clinical significance and the relationship between HER2 gene amplification and expression. PLoS One 9, e98528. https://doi.org/10.1371/journal.pone.0098528 (2014).
Li, N., Bu, X., Wu, P., Wu, P. & Huang, P. The, “HER2-PI3K/Akt-FASN Axis” regulated malignant phenotype of colorectal cancer cells. Lipids 47, 403–411. https://doi.org/10.1007/s11745-011-3649-7 (2012).
Casak, S. J. et al. FDA approval summary: Tucatinib with trastuzumab for advanced unresectable or metastatic, chemotherapy refractory, HER2-positive RAS wild-type colorectal cancer. Clin. Cancer Res. 29, 4326–4330. https://doi.org/10.1158/1078-0432.CCR-23-1041 (2023).
Afrasanie, V. A. et al. KRAS, NRAS, BRAF, HER2 and microsatellite instability in metastatic colorectal cancer—Practical implications for the clinician. Radiol. Oncol. 53, 265–274. https://doi.org/10.2478/raon-2019-0033 (2019).
Alexander, D. et al. Postsurgical disparity in survival between African Americans and Caucasians with colonic adenocarcinoma. Cancer 101, 66–76. https://doi.org/10.1002/cncr.20337 (2004).
Gong, J., Cho, M., Sy, M., Salgia, R. & Fakih, M. Molecular profiling of metastatic colorectal tumors using next-generation sequencing: A single-institution experience. Oncotarget 8, 42198–42213. https://doi.org/10.18632/oncotarget.15030 (2017).
Huang, H., Wang, Z., Li, Y., Zhao, Q. & Niu, Z. Amplification of the human epidermal growth factor receptor 2 (HER2) gene is associated with a microsatellite stable status in Chinese gastric cancer patients. J. Gastrointest. Oncol. 12, 377–387. https://doi.org/10.21037/jgo-21-47 (2021).
Benson, A. B. et al. Colon Cancer, Version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 19, 329–359. https://doi.org/10.6004/jnccn.2021.0012 (2021).
Ingold Heppner, B. et al. HER2/neu testing in primary colorectal carcinoma. Br. J. Cancer 111, 1977–1984. https://doi.org/10.1038/bjc.2014.483 (2014).
Missiaglia, E. et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 25, 1995–2001. https://doi.org/10.1093/annonc/mdu275 (2014).
Wu, S. W., Ma, C. C. & Li, W. H. Does overexpression of HER-2 correlate with clinicopathological characteristics and prognosis in colorectal cancer? Evidence from a meta-analysis. Diagn. Pathol. 10, 144. https://doi.org/10.1186/s13000-015-0380-3 (2015).
Shin, I. Y. et al. The expression of multiple proteins as prognostic factors in colorectal cancer: Cathepsin D, p53, COX-2, epidermal growth factor receptor, C-erbB-2, and Ki-67. Gut Liver 8, 13–23. https://doi.org/10.5009/gnl.2014.8.1.13 (2014).
McKay, J. A. et al. c-erbB-2 is not a major factor in the development of colorectal cancer. Br. J. Cancer 86, 568–573. https://doi.org/10.1038/sj.bjc.6600127 (2002).
Cancer Genome Atlas Nature. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. https://doi.org/10.1038/nature11252 (2012).
Salem, M. E. et al. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget 8, 86356–86368. https://doi.org/10.18632/oncotarget.21169 (2017).
Baiocchi, G. et al. ErbB family immunohistochemical expression in colorectal cancer patients with higher risk of recurrence after radical surgery. Int. J. Colorectal Dis. 24, 1059–1068. https://doi.org/10.1007/s00384-009-0702-6 (2009).
Lu, Y. et al. Expression of EGFR, HER2 predict lymph node metastasis (LNM)-associated metastasis in colorectal cancer. Cancer Biomark. 11, 219–226. https://doi.org/10.3233/CBM-2012-00282 (2012).
Kavanagh, D. O. et al. Is overexpression of HER-2 a predictor of prognosis in colorectal cancer?. BMC Cancer 9, 1. https://doi.org/10.1186/1471-2407-9-1 (2009).
Meric-Bernstam, F. et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 20, 518–530. https://doi.org/10.1016/S1470-2045(18)30904-5 (2019).
Guo, Y. et al. Genomic alterations of NTRK, POLE, ERBB2, and microsatellite instability status in Chinese patients with colorectal cancer. Oncologist 25, e1671–e1680. https://doi.org/10.1634/theoncologist.2020-0356 (2020).
Sun, C. et al. Treatment of advanced BRAF-mutated colorectal cancer: Where we are and where we are going. Clin. Colorectal Cancer 21, 71–79. https://doi.org/10.1016/j.clcc.2022.01.009 (2022).
Rosati, G., Aprile, G., Basile, D. & Avallone, A. Perspectives in the treatment of RAS or BRAF mutated metastatic colorectal cancer patients. Front. Oncol. 11, 602596. https://doi.org/10.3389/fonc.2021.602596 (2021).
Corcoran, R. B. et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2, 227–235. https://doi.org/10.1158/2159-8290.CD-11-0341 (2012).
Pietrantonio, F. et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 51, 587–594. https://doi.org/10.1016/j.ejca.2015.01.054 (2015).
Imai, K. & Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 29, 673–680. https://doi.org/10.1093/carcin/bgm228 (2008).
Jensen, L. H., Lindebjerg, J., Byriel, L., Kolvraa, S. & Cruger, D. G. Strategy in clinical practice for classification of unselected colorectal tumours based on mismatch repair deficiency. Colorectal Dis. 10, 490–497. https://doi.org/10.1111/j.1463-1318.2007.01378.x (2008).
Ahmed, M. Colon cancer: A clinician’s perspective in 2019. Gastroenterol. Res. 13, 1–10. https://doi.org/10.14740/gr1239 (2020).
Prior, I. A., Hood, F. E. & Hartley, J. L. The frequency of RAS mutations in cancer. Cancer Res. 80, 2969–2974. https://doi.org/10.1158/0008-5472.CAN-19-3682 (2020).
Negri, F., Bottarelli, L., de’Angelis, G. L. & Gnetti, L. KRAS: A druggable target in colon cancer patients. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23084120 (2022).
Ciardiello, F. et al. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J. Clin. 72, 372–401. https://doi.org/10.3322/caac.21728 (2022).
Schirripa, M. et al. Role of NRAS mutations as prognostic and predictive markers in metastatic colorectal cancer. Int. J. Cancer 136, 83–90. https://doi.org/10.1002/ijc.28955 (2015).
Raghav, K. et al. Validation of HER2 amplification as a predictive biomarker for anti-epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. JCO Precis. Oncol. 3, 1–13. https://doi.org/10.1200/PO.18.00226 (2019).
Malla, M., Pedersen, K. S. & Parikh, A. R. Updates in the treatment of metastatic colorectal cancer. J. Natl. Compr. Cancer Netw. 21, 567–571. https://doi.org/10.6004/jnccn.2023.5012 (2023).
Nakayama, M. & Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 11, 267–276. https://doi.org/10.1093/jmcb/mjy075 (2019).
Iacopetta, B. TP53 mutation in colorectal cancer. Hum. Mutat. 21, 271–276. https://doi.org/10.1002/humu.10175 (2003).
Russo, A. et al. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: Influence of tumor site, type of mutation, and adjuvant treatment. J. Clin. Oncol. 23, 7518–7528. https://doi.org/10.1200/JCO.2005.00.471 (2005).
Shah, S. P. et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486, 395–399. https://doi.org/10.1038/nature10933 (2012).
Silwal-Pandit, L. et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin. Cancer Res. 20, 3569–3580. https://doi.org/10.1158/1078-0432.CCR-13-2943 (2014).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb Perspect. Biol. 2, a001008. https://doi.org/10.1101/cshperspect.a001008 (2010).
Jessup, J. M. et al. Colon and Rectum. In AJCC Cancer Staging Manual 8th edn (eds Amin, M. et al.) 251–274 (Springer Nature, 2017).
Hofmann, M. et al. Assessment of a HER2 scoring system for gastric cancer: Results from a validation study. Histopathology 52, 797–805. https://doi.org/10.1111/j.1365-2559.2008.03028.x (2008).
Ruschoff, J. et al. HER2 testing in gastric cancer: A practical approach. Mod. Pathol. 25, 637–650. https://doi.org/10.1038/modpathol.2011.198 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. https://doi.org/10.1101/gr.107524.110 (2010).
Cingolani, P. et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 3, 35. https://doi.org/10.3389/fgene.2012.00035 (2012).
Liu, X., Wu, C., Li, C. & Boerwinkle, E. dbNSFP v30: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 37, 235–241. https://doi.org/10.1002/humu.22932 (2016).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. https://doi.org/10.1038/nbt.1754 (2011).
Ross, J. S. et al. Targeting HER2 in colorectal cancer: The landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer 124, 1358–1373. https://doi.org/10.1002/cncr.31125 (2018).
Acknowledgements
This work was supported by a research grant from Jeju National University Hospital in 2021 (JNUH2021). We would like to thank Ms. So-Yeon Park (Histotechnician) for performing immunohistochemistry and DISH and managing its quality control.
Author information
Authors and Affiliations
Contributions
S.L. designed study, collected data and involved study management, statistical analysis, and data interpretation. S.L. also wrote the first draft of the manuscript. H.O. critically reviewed the first draft of the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, S.M., Oh, H. RAS/RAF mutations and microsatellite instability status in primary colorectal cancers according to HER2 amplification. Sci Rep 14, 11432 (2024). https://doi.org/10.1038/s41598-024-62096-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-62096-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
