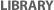Figure 3: Pedigree of a South African family with Beckwith-Weidemann syndrome.
Figure 3: Pedigree of a South African family with Beckwith-Weidemann syndrome.















« Prev Next »
Many illnesses are rooted in our genetic code. Others, however, are caused by changes in how our genes are regulated, rather than changes in the specific nucleotide sequence itself. These diseases often stem from epigenetic mechanisms gone astray.
The term "epigenetic" refers to those characteristics that are propagated from cell to cell by some means other than base-pairing, which makes them different from classic Mendelian traits. Usually, we inherit two copies of a gene, one from each parent, and both copies actively shape how we develop. However, if one of those copies is epigenetically "turned off" and the remaining working gene is deleted or severely mutated, then a person can be debilitated. In this case, the person has a good copy of the needed gene, but this gene cannot act because it has been turned off by epigenetic mechanisms.
But how, exactly, can one parent's contributed gene be shut off? Called genetic imprinting, the turning off of one parent's gene is thought to occur during the formation of an egg or a sperm cell. Part of the mechanism that turns off the parent's gene is methylation (i.e., the attachment of a methyl group) of that parent's DNA. Over the years, a number of diseases and disorders have been linked to this sort of genetic imprinting, including Angelman syndrome, Prader-Willi syndrome, and Beckwith-Wiedemann syndrome.
Angelman and Prader-Willi Syndromes

Figure 1: Andrew, a child with Angelman syndrome.
© 2008 Angel Syndrome Foundation, Inc. Courtesy of the Heinzmann family. All rights reserved. 
Despite the widespread prevalence of Angelman syndrome, the chromosomal abnormality underlying this disease remained unknown for several decades following Angelman's initial report. Eventually, however, thanks to advances in research methods, a deleted area in chromosome 15 was detected (Figure 2; Magenis et al., 1987); this deletion occurs in about 70% of Angelman patients. Although the deletion appears to be extremely small (and therefore hard to detect), it is quite significant. This deletion, which is believed to be about 4 million base pairs in length, includes a gene involved in the ubiquitin pathway known as UBE3A, for which the maternal gene is normally expressed while the paternal gene is silenced. The loss of that maternal contribution, by either deletion (as in 70% of Angelman patients) or other genetic abnormalities (in the other 30% of cases), results in the phenotypic symptoms observed in Angelman syndrome. Here, the paternal gene cannot fill in the blank because it has been turned off by imprinting (Figure 2). Note that there have also been rare cases in which the maternal allele has been lost completely, resulting in two copies of this gene from the father (a condition known as uniparental disomy). Moreover, if the maternal locus is errantly imprinted, this too can result in Angelman syndrome.
The region of chromosome 15 that is involved in Angelman syndrome also
contains another important gene that is imprinted the other way. In this
case, when the paternal contribution is lost, the result is a condition
known as Prader-Willi syndrome (PWS). PWS patients suffer from extreme
feeding problems, including hyperphagia, or extreme, insatiable appetite
and obsession with food. Affected children are also developmentally
delayed for motor skills due to decreased muscle tone. As previously
mentioned, PWS is caused by the loss of other genes in this same region,
including SNRPN. This gene, normally active from the paternal
copy of chromosome 15, encodes a component of mRNA splicing.
In short, imprinting of the same region on chromosome 15 has been implicated for both Angelman and Prader-Willi syndromes. However, it is the loss of the maternal contribution that is linked to Angelman syndrome and the loss of the paternal contribution that is linked to PWS. In each case, the presence of a second good copy of the gene(s) on the methylated, tightly packed copy of chromosome 15 is of no use in correcting the defect.
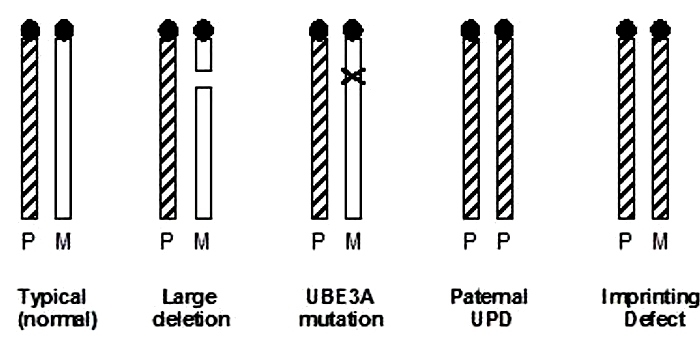
Figure 2: Genetic mechanisms leading to Angelman syndrome.
The rectangles represent chromosome 15. Hatched chromosomes have a paternal pattern of gene functioning and DNA methylation; open chromosomes have a maternal pattern. Angelman syndrome is caused by a large deletion of the region of the maternal chromosome that contains UBE3A, or by a DNA sequence change (mutation) in the UBE3A gene inherited from the mother. The condition can also be caused by inheritance of two normal copies of UBE3A from the father, with no copy inherited from the mother. Other AS individuals inherited chromosome 15 from their mother, but that chromosome had the paternal pattern of gene function and DNA methylation.
© 2008 Angel Syndrome Foundation, Inc. All rights reserved.
Beckwith-Wiedemann Syndrome
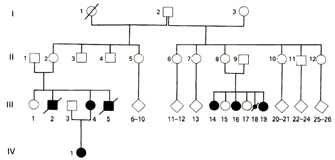
© 1992 BMJ Publishing Group, Ltd. Viljoen, D. et. al. Evidence for paternal imprinting in familial Beckwith-Wiedemann syndrome. Journal of Medical Genetics 29, 221-225 (1992). All rights reserved.
Determining the exact genetic nature of BWS was not a simple process. The small numbers of affected individuals made it difficult for investigators to definitively show that imprinting was responsible. Eventually, some investigators linked the condition to chromosome region 11p15.5. Specifically, Henry et al. (1991) collected genotype information from BWS patients and discovered that several of them were missing evidence of a maternal allele at specific loci in this region. Instead, both alleles came from the patients' fathers, making this condition an example of uniparental paternal disomy (Henry et al., 1991). Moreover, when the investigators examined the genotypes of the tumors common in BWS patients, they discovered additional evidence for uniparental disomy in the tumor cells. These data suggested the possibility that paternal imprinting was responsible for BWS. With this new information in hand, the researchers were able to reexamine medical history data from different BWS families in order to determine whether paternal imprinting indeed fit the clinical data. Multiple different inheritance patterns had previously been suggested, including autosomal recessive inheritance by both Beckwith and Wiedemann. However, after examining 27 different families with BWS, including the South African family shown in Figure 3 (Viljoen & Ramesar, 1992), the investigators noted that a majority of the BWS cases could indeed be explained by paternal imprinting.
This new pedigree showed an unaffected male with two grandchildren with the disorder. The two grandchildren had different mothers. This pedigree strongly suggested that the man's genes were involved, but they were not expressed until after passing through the female line. In this example, in the oldest generation (I), the imprint in individual I.2 was normal. Moreover, the imprint had gone awry in the germ line of only a subset of this individual's children (generation II, individuals II.2 and II.8). These females were themselves healthy but had a 50% chance of passing the incorrectly imprinted gene to their children. In fact, this happened to both of these women, resulting in affected children in the third generation (shown in dark circles and squares).
Through this sort of analysis, Beckwith-Wiedemann syndrome was determined to be the result of mutations in chromosomal region 11p15.5. Interestingly, a number of genes in this area have all been linked to Beckwith-Wiedemann, including H19 and Igf2. Moreover, beyond causing abnormal growth both in the uterus and after birth, genetic imprinting also plays a role in the development of cancers in Beckwith-Wiedemann syndrome. Both decreased expression of tumor suppressor genes and overexpression of oncogenes may be involved. Indeed, particular patterns of genetic imprinting in Beckwith-Wiedemann syndrome may be associated with the development of particular types of tumors. As the genetic pathways involved in BWS continue to be elucidated, researchers hope this knowledge will further inform risk assessment, early detection, and diagnosis of cancers in Beckwith-Wiedemann patients, thereby improving treatment methods and outcomes.
References and Recommended Reading
Angelman, H. "Puppet" children: A report of three cases. Developmental Medicine and Child Neurology 7, 681–688 (1965)
Henry, I., et al. Uniparental paternal disomy in a genetic cancer-predisposing syndrome. Nature 351, 665–667 (1991) doi:10.1038/351665a0 (link to article)
Magenis, R. E., et al. Is Angelman syndrome an alternate result of del(15)(q11q13)? American Journal of Medical Genetics 28, 829–838 (1987)
Viljoen, D., & Ramesar, R. Evidence for paternal imprinting in familial Beckwith-Wiedemann syndrome. Journal of Medical Genetics 29, 221–225 (1992)