Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain
the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in
Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles
and JavaScript.
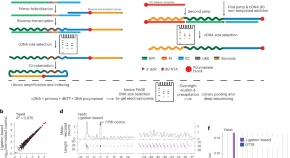
A comprehensive redevelopment of the ribosome profiling workflow involves improved nuclease treatment and sequencing library preparation, enabling richer and more accurate translatome profiling with lower input and fewer technical hurdles.
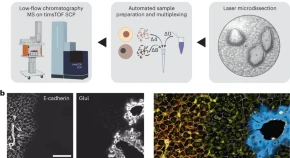
Single-cell Deep Visual Proteomics integrates imaging, cell segmentation, laser microdissection and multiplexed mass spectrometry for spatial single-cell proteomics measurements in complex tissues.
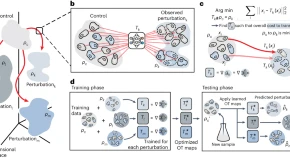
CellOT combines the benefits of optimal transport and input convex neural architectures to directly learn and uncover maps between control and perturbed cell states at the single-cell level.
A deep learning approach bypasses iterative trials associated with sensorless adaptive optics to compensate for wavefront deformations when imaging biological specimens, enabling improved deep tissue localization microscopy.
Guided sparse factor analysis (GSFA) is a powerful statistical framework to detect changes in gene expression as a result of perturbations in single-cell CRISPR screening.
D-LMBmap is a fully automated pipeline for mesoscale connectomics including deep-learning modules for axon segmentation, brain region segmentation and whole-brain registration. D-LMBmap works accurately across cell types and modalities.
AlteredPQR is a software tool, available as an R package, to infer remodeling of protein functional modules from whole-cell or tissue lysate proteomic measurements.
A Ni2+-modified MspA nanopore construct can unambiguously discriminate the 20 proteogenic amino acids as well as several post-translational modifications.
EzMechanism is a tool for automated prediction of the catalytic mechanisms of enzymes using their three-dimensional structures and chemical reactions as input.
veloVI enhances RNA velocity analysis with uncertainty quantification and extensibility by deep generative modeling of gene-specific transcriptional dynamics.
The GelMap workflow adds a fluorescent grid into samples before expansion, allowing for precise determination of expansion factor and subsequent deformation correction in ExM. GelMap works with diverse samples and expansion methods.
Montage parallel array cryo-tomography adopts principles of montage tomography via regular array beam-image-shift montage acquisition and is robust for imaging large fields of view while retaining high-resolution structural information in cryo-electron tomography.
The Clivias are a series of small, monomeric fluorescent RNAs that emit with a large Stokes shift in the orange–red. They enable multiplexed RNA imaging in live cells and BRET-based detection of protein–RNA interactions in mice.
Statistically unbiased prediction utilizing spatiotemporal information in imaging data (SUPPORT) is a self-supervised deep learning approach to accurately denoise voltage and calcium imaging data while preserving true dynamic signals.
This work introduces a wet lab and computational pipeline, Napu, for small variant calling and de novo assembly of Nanopore sequencing data, which leads to comparable performances to short-read sequencing and allows for large-scale long-read sequencing projects.
This paper presents an improved approach for mapping single-cell RNA-seq reads with optimized transcriptomic references, which markedly recovers previously missing gene expression data.
Multielement Z-tag X-ray fluorescence (MEZ-XRF) offers a new avenue for nondestructive and highly multiplexed tissue imaging and operates from the nanometer to whole-tissue scale, unlocking new biological observations.