
Abstract
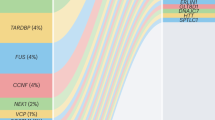
Considerable progress has been made in unraveling the genetic etiology of amyotrophic lateral sclerosis (ALS), the most common form of adult-onset motor neuron disease and the third most common neurodegenerative disease overall. Here we review genes implicated in the pathogenesis of motor neuron degeneration and how this new information is changing the way we think about this fatal disorder. Specifically, we summarize current literature of the major genes underlying ALS, SOD1, TARDBP, FUS, OPTN, VCP, UBQLN2, C9ORF72 and PFN1, and evaluate the information being gleaned from genome-wide association studies. We also outline emerging themes in ALS research, such as next-generation sequencing approaches to identify de novo mutations, the genetic convergence of familial and sporadic ALS, the proposed oligogenic basis for the disease, and how each new genetic discovery is broadening the phenotype associated with the clinical entity we know as ALS.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others
References
Rowland, L.P. & Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700 (2001).
Rosen, D.R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993). This study was the first to identify a genetic cause of familial ALS.
Sreedharan, J. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 (2008). This study identified mutations in TARDBP , which encodes the TDP-43 protein, as a cause of familial ALS.
Deng, H.X. et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215 (2011).
Johnson, J.O. et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864 (2010). This study was the first to apply exome sequencing to identify a genetic cause of familial ALS and represents an initial step in unraveling the genetic overlap between ALS and FTD.
Wu, C.H. et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503 (2012).
Renton, A.E. et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). Refs. 7 and 8 identified the hexanucleotide repeat expansion in C9ORF72 as a major cause of familial and sporadic ALS and FTD.
Maruyama, H. et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226 (2010).
Fecto, F. et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446 (2011).
Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 6, 37–46 (2006).
Chiò, A. et al. Prevalence of SOD1 mutations in the Italian ALS population. Neurology 70, 533–537 (2008).
Cudkowicz, M.E. et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol. 41, 210–221 (1997).
Andersen, P.M. et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain 119, 1153–1172 (1996).
Turner, M.R. et al. Distinct cerebral lesions in sporadic and 'D90A' SOD1 ALS: studies with [11C]flumazenil PET. Brain 128, 1323–1329 (2005).
Pasinelli, P. et al. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43, 19–30 (2004).
Ludolph, A.C. et al. Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: report on the 142nd ENMC international workshop. Amyotroph. Lateral Scler. 8, 217–223 (2007).
Mackenzie, I.R. et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434 (2007).
Miller, T.M. et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442 (2013). This study was the first report of gene therapy using antisense oligonucleotides in ALS patients.
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). This seminal paper identified TDP-43 as a major component of the ubiquitin-positive neuronal inclusions that are the neuropathological hallmark of ALS and FTD.
Chiò, A. et al. Amyotrophic lateral sclerosis-frontotemporal lobar dementia in 3 families with p.Ala382Thr TARDBP mutations. Arch. Neurol. 67, 1002–1009 (2010).
Chiò, A. et al. Extensive genetics of ALS: a population-based study in Italy. Neurology 79, 1983–1989 (2012).
Chiò, A. et al. Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch. Neurol. 68, 594–598 (2011).
Lagier-Tourenne, C. et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497 (2012).
Kwiatkowski, T.J. Jr. et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208 (2009).
Vance, C. et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211 (2009).
Kabashi, E. et al. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 7, e1002214 (2011).
Rezaie, T. et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295, 1077–1079 (2002).
Albagha, O.M. et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget's disease of bone. Nat. Genet. 42, 520–524 (2010).
Kachaner, D., Génin, P., Laplantine, E. & Weil, R. Toward an integrative view of Optineurin functions. Cell Cycle 11, 2808–2818 (2012).
Koppers, M. et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 33, 837.e7–837.e13 (2012).
DeJesus-Hernandez, M. et al. Novel p.Ile151Val mutation in VCP in a patient of African American descent with sporadic ALS. Neurology 77, 1102–1103 (2011).
Watts, G.D. et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381 (2004).
Kim, H.J. et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 (2013).
Bartolome, F. et al. Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64 (2013).
Daoud, H. et al. UBQLN2 mutations are rare in French and French-Canadian amyotrophic lateral sclerosis. Neurobiol. Aging 33, 2230 e1–2230.e5 (2012).
Majounie, E. et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330 (2012).
Lesage, S. et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 136, 385–391 (2013).
Majounie, E. et al. Repeat expansion in C9ORF72 in Alzheimer's disease. N. Engl. J. Med. 366, 283–284 (2012).
Chiò, A. et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 135, 784–793 (2012).
Beck, J. et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 92, 345–353 (2013).
Xi, Z. et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 92, 981–989 (2013).
Fratta, P. et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2, 1016 (2012).
Mori, K. et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013).
Todd, P.K. et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455 (2013).
Laurin, N., Brown, J.P., Morissette, J. & Raymond, V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588 (2002).
Teyssou, E. et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 125, 511–522 (2013).
Rubino, E. et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79, 1556–1562 (2012).
Lattante, S., Le Ber, I., Camuzat, A., Brice, A. & Kabashi, E. Mutations in the PFN1 gene are not a common cause in patients with amyotrophic lateral sclerosis and frontotemporal lobar degeneration in France. Neurobiol. Aging 34, 1709 e1–1709.e2 (2013).
Hadano, S. et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29, 166–173 (2001).
Eymard-Pierre, E. et al. Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am. J. Hum. Genet. 71, 518–527 (2002).
Chen, Y.Z. et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 74, 1128–1135 (2004).
Duquette, A. et al. Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann. Neurol. 57, 408–414 (2005).
Chow, C.Y. et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 84, 85–88 (2009).
Mitchell, J. et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc. Natl. Acad. Sci. USA 107, 7556–7561 (2010).
Puls, I. et al. Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456 (2003).
Farrer, M.J. et al. DCTN1 mutations in Perry syndrome. Nat. Genet. 41, 163–165 (2009).
Nishimura, A.L. et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831 (2004).
Landers, J.E. et al. New VAPB deletion variant and exclusion of VAPB mutations in familial ALS. Neurology 70, 1179–1185 (2008).
Skibinski, G. et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808 (2005).
Parkinson, N. et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077 (2006).
Cox, L.E. et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 5, e9872 (2010).
Greenway, M.J. et al. A novel candidate region for ALS on chromosome 14q11.2. Neurology 63, 1936–1938 (2004).
Greenway, M.J. et al. ANG mutations segregate with familial and 'sporadic' amyotrophic lateral sclerosis. Nat. Genet. 38, 411–413 (2006).
Millecamps, S. et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J. Med. Genet. 47, 554–560 (2010).
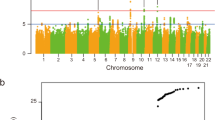
Schymick, J.C. et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 6, 322–328 (2007). This was the first genome-wide association study of ALS.
Dunckley, T. et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 357, 775–788 (2007).
van Es, M.A. et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 6, 869–877 (2007).
Cronin, S. et al. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum. Mol. Genet. 17, 768–774 (2008).
van Es, M.A. et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 40, 29–31 (2008).
Chiò, A. et al. A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 1524–1532 (2009).
Landers, J.E. et al. Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 106, 9004–9009 (2009).
van Es, M.A. et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 41, 1083–1087 (2009).
Traynor, B.J. et al. Kinesin-associated protein 3 (KIFAP3) has no effect on survival in a population-based cohort of ALS patients. Proc. Natl. Acad. Sci. USA 107, 12335–12338 (2010).
Laaksovirta, H. et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 9, 978–985 (2010). This was the first genome-wide association study to identify the chromosome 9p21 association signal in the founder Finnish population.
Shatunov, A. et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 9, 986–994 (2010).
Kwee, L.C. et al. A high-density genome-wide association screen of sporadic ALS in US veterans. PLoS ONE 7, e32768 (2012).
Ahmeti, K.B. et al. Age of onset of amyotrophic lateral sclerosis is modulated by a locus on 1p34.1. Neurobiol. Aging 34, 357 e7–357.e19 (2013).
Deng, M. et al. Genome-wide association analyses in Han Chinese identify two new susceptibility loci for amyotrophic lateral sclerosis. Nat. Genet. 45, 697–700 (2013).
Fogh, I. et al. No association of DPP6 with amyotrophic lateral sclerosis in an Italian population. Neurobiol. Aging 32, 966–967 (2011).
Daoud, H. et al. Analysis of the UNC13A gene as a risk factor for sporadic amyotrophic lateral sclerosis. Arch. Neurol. 67, 516–517 (2010).
Van Hoecke, A. et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 18, 1418–1422 (2012).
Schymick, J.C., Talbot, K. & Traynor, B.J. Genetics of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 16 (spec. no. R2): R233–R242 (2007).
Gwinn, K. et al. Amyotrophic lateral sclerosis: an emerging era of collaborative gene discovery. PLoS ONE 2, e1254 (2007).
Hollingworth, P. et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435 (2011).
Nalls, M.A. et al. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 377, 641–649 (2011).
Cronin, S., Hardiman, O. & Traynor, B.J. Ethnic variation in the incidence of ALS: a systematic review. Neurology 68, 1002–1007 (2007).
Littler, M. & Morton, N.E. Segregation analysis of peripheral neurofibromatosis (NF1). J. Med. Genet. 27, 307–310 (1990).
Ivanchuk, S.M., Myers, S.M., Eng, C. & Mulligan, L.M. De novo mutation of GDNF, ligand for the RET/GDNFR-alpha receptor complex, in Hirschsprung disease. Hum. Mol. Genet. 5, 2023–2026 (1996).
Alexander, M.D. et al. “True” sporadic ALS associated with a novel SOD-1 mutation. Ann. Neurol. 52, 680–683 (2002). This study was the first to identify a de novo mutation in any ALS gene, thereby demonstrating that at least a portion of sporadic disease was due to spontaneous mutations.
Chiò, A. et al. A de novo missense mutation of the FUS gene in a “true” sporadic ALS case. Neurobiol. Aging 32, 553.e23–553.e26 (2011).
DeJesus-Hernandez, M. et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum. Mutat. 31, E1377–E1389 (2010).
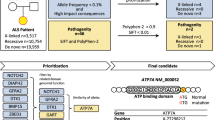
Chesi, A. et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat. Neurosci. 16, 851–855 (2013). This was the first study to apply exome sequencing of trios to identify de novo mutations as a possible cause of sporadic ALS.
Turner, M.R. et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322 (2013).
Chiò, A. et al. Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J. Neurol. Neurosurg. Psychiatry 10.1136/jnnp-2013-305546 (6 July 2013).
van Blitterswijk, M. et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 21, 3776–3784 (2012).
Chiò, A. et al. ALS/FTD phenotype in two Sardinian families carrying both C9ORF72 and TARDBP mutations. J. Neurol. Neurosurg. Psychiatry 83, 730–733 (2012).
Elden, A.C. et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075 (2010).
Van Damme, P. et al. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology 76, 2066–2072 (2011).
van Es, M.A. et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann. Neurol. 70, 964–973 (2011).
Acknowledgements
This work was supported in part by the Intramural Research Programs of the US National Institutes of Health, National Institute on Aging (Z01-AG000949-02) and National Institute of Neurological Disorders and Stroke. The work was also supported by the Packard Center for ALS Research at Hopkins (B.J.T.), the ALS Association (B.J.T., A.C.), Microsoft Research (B.J.T.), AriSLA (B.J.T., A.C.), the Italian Health Ministry (Ricerca Sanitaria Finalizzata 2007 to A.C.), Fondazione Vialli e Mauro ONLUS (A.C.), Federazione Italiana Giuoco Calcio (A.C., B.J.T.), Compagnia di San Paolo (A.C.) and the European Community's Health Seventh Framework Programme under grant agreement 259867 (A.C.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
B.J.T. has a patent pending on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9ORF72.
Rights and permissions
About this article
Cite this article
Renton, A., Chiò, A. & Traynor, B. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17, 17–23 (2014). https://doi.org/10.1038/nn.3584
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.3584
This article is cited by
-
Beyond C9orf72: repeat expansions and copy number variations as risk factors of amyotrophic lateral sclerosis across various populations
BMC Medical Genomics (2024)
-
Choroid plexus enlargement in amyotrophic lateral sclerosis patients and its correlation with clinical disability and blood-CSF barrier permeability
Fluids and Barriers of the CNS (2024)
-
Genetics screening in an Italian cohort of patients with Amyotrophic Lateral Sclerosis: the importance of early testing and its implication
Journal of Neurology (2024)
-
Oxytosis/Ferroptosis in Neurodegeneration: the Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4)
Molecular Neurobiology (2024)
-
Loss of TDP-43 mediates severe neurotoxicity by suppressing PJA1 gene transcription in the monkey brain
Cellular and Molecular Life Sciences (2024)
