
Abstract
Global climate fluctuated considerably throughout the Pliocene and Pleistocene, influencing the evolutionary history of a wide range of species. Using both mitochondrial sequences and microsatellites, we have investigated the evolutionary consequences of such environmental fluctuation for the patterns of genetic variation in the common warthog, sampled from 24 localities in Africa. In the sample of 181 individuals, 70 mitochondrial DNA haplotypes were identified and an overall nucleotide diversity of 4.0% was observed. The haplotypes cluster in three well-differentiated clades (estimated net sequence divergence of 3.1–6.6%) corresponding to the geographical origins of individuals (i.e. eastern, western and southern African clades). At the microsatellite loci, high polymorphism was observed both in the number of alleles per locus (6–21), and in the gene diversity (in each population 0.59–0.80). Analysis of population differentiation indicates greater subdivision at the mitochondrial loci (FST=0.85) than at nuclear loci (FST=0.20), but both mitochondrial and nuclear loci support the existence of the three warthog lineages. We interpret our results in terms of the large-scale climatic fluctuations of the Pleistocene.
Similar content being viewed by others

Introduction
Global climate has fluctuated greatly during the past 3 million years, leading to great changes in the ranges of many species in all parts of the world. The fossil record in Europe and North America indicates that, during this period, some species went extinct over large parts of their range, some dispersed to new locations and some survived in refugia from which they later dispersed. Cycles of retreat and dispersal might have occurred repeatedly (Coope, 1994; Paulo et al, 2001). In Africa too, cyclic climatic fluctuations had dramatic effects on the geographical ranges of the main vegetation types and their associated animals (Hamilton, 1982; deMenocal, 1995). Recent DNA studies of some large African mammals show how climatic fluctuations affected their distribution and genomic divergence. For some species such as the impala (Aepyceros melampus), greater kudu (Tragelaphus strepsiceros) and wildebeest (Connochaetes taurinus), patterns of genetic diversity suggest a colonization process from southern Africa where isolated populations might have survived during adverse times (Arctander et al, 1999; Nersting and Arctander, 2001). In others such as the topi (Damaliscus lunatus) and hartebeest (Alcelaphus buselaphus), the patterns suggest that they became extinct except in a few places in southern and eastern Africa from where they expanded when environmental conditions improved (Arctander et al, 1999; Flagstad et al, 2001).
Climatic fluctuations have probably influenced the genetic structure of the common warthog (Phacochoerus africanus) as well. The warthog is the most wide spread extant wild pig species. It occurs practically everywhere in Africa, except in arid regions and in tropical forests. Owing to fragmentation of habitats and conflict with agriculture for land use, warthogs, like other large mammals, are now restricted to protected areas. Up to 10 different subspecies in the warthog were originally named on the basis of cranial characters (reviewed in Meester and Setzer, 1971). Kingdon (1989) however dismissed the recognition of subspecies. Recently, only four subspecies were provisionally recognized (Grubb, 1993): P. africanus massaicus (inhabiting eastern Africa), P. africanus sundevallii (southern Africa) P. africanus africanus (western Africa) and P. africanus aeliani (Eritrea and Ethiopia).
There is an extensive fossil record of phacochoerine pigs (open country species resembling the warthog) in the middle Pleistocene (0.8 myr). The earliest recorded fossil resembling the modern common warthog is P. antiquus, whose First Appearance Datum (FAD) is 0.78 myr (White and Harris, 1977; White, 1995). Fossil remains of the modern warthog are rare in the middle Pleistocene but expanded in the Upper Pleistocene (0.4 myr) when the other phacochoerine pigs were becoming extinct (Kingdon, 1989). In this paper, we use mitochondrial nucleotide sequence variation together with allele length variation at microsatellite loci to investigate the effects of past environmental fluctuations on patterns of genetic variation in the common warthog.
Materials and methods
Sample collection and preservation
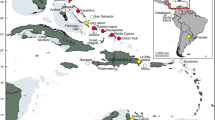
In total, 181 samples were collected of which 33 were obtained from western Africa (Ghana and Cameroon), 27 from southern Africa (Zambia, Zimbabwe and Namibia) and 121 from eastern Africa (Uganda, Kenya and Tanzania) (Figure 1). It was difficult to obtain samples and as a result, sample sizes vary extensively among localities. Analyses performed at population level were restricted to localities with at least six individuals (see Table 1 for abbreviations and sample sizes). A mitochondrial DNA sequence of the domestic pig, Sus scrofa (Kim et al, 2002) was retrieved from GenBank (accession no. AF276937) and used as an outgroup. Samples were obtained either as small skin biopsies from free-ranging individuals, tissues from hunters or mucosal lining on fresh warthog dung (only 30 samples were obtained this way). All samples were preserved in 25% dimethylsulfoxide saturated with sodium chloride (Amos and Hoelzel, 1991) and stored at ambient temperature in the field and at −80°C in the laboratory.

Map of Africa showing the distribution of the common warthog and the geographical locations of the sampling localities (some localities with one individual each are not shown). The four provisional subspecies according to Grubb (1993) (P. africanus massaicus, P. africanus sundevallii, P. africanus africanus and P. africanus aeliani) are indicated with different patterns.
DNA extraction
Total genomic DNA was extracted from the samples using standard procedures either involving treatment with sodium dodecyl sulfate and proteinase K, and subsequent phenol/chloroform extraction (Sambrook et al, 1989) or by use of the Dneasy tissue kit (QIAGEN) following the manufacturers' protocol.
Mitochondrial control region
Amplification and sequencing: An approximately 370 bp-long fragment of the variable 5′ part of the mtDNA control region (d-loop) was polymerase chain reaction (PCR) amplified using the primers MT4 (5′CCTCCCTAAGACTCAAGGAAG3′) (Arnason et al, 1993) and PeaR (5′AGTTCATAATTGAAACCCCCA3′). PeaR is located upstream of the d-loop and was specifically designed to amplify the 5′ part of warthog d-loop in conjunction with MT4. Symmetrical PCR amplifications were carried out in 50-μl reaction volumes containing 10 ng of total genomic DNA, 50 pmol of each of the primers, 5 × PCR reaction buffer (Boehringer Mannheim GmbH), 50 pmol dNTPs and 1 U of Taq polymerase (Boehringer Mannheim GmbH). We used one cycle of denaturation at 94°C for 5 min followed by 30 cycles of denaturation at 94°C for 1 min, annealing at 52°C for 1 min, and extension at 72°C for 1 min 30 s. One primer was 5′-end biotinylated, and the double-stranded PCR product was separated into single strands using streptavidin-coated paramagnetic beads (DYNAL®). Single-stranded DNA was dissolved in distilled water and used as the template for sequencing by the dideoxy chain-termination method (Sanger et al, 1977) using the sequenase kit version 2.0 (Amersham Pharmacia Biotech, Inc.), [α-35]-dATP (Amersham Pharmacia Biotech, Inc.) and a nonbiotinylated primer complementary to the template. Both strands were sequenced. Products of the sequencing reaction were electrophoresed in 6% polyacrylamide/7 M-urea gel. The gel was fixed, dried exposed on a Kodak film for 24–48 h and read manually.
Sequence analysis
Phylogenetic analysis Sequences were aligned by eye using the program SeqApp version 1.9 (Gilbert, 1993). Insertions/deletions were introduced so as to minimize transversions. Phylogenetic relationships between haplotypes were estimated in the following two ways:
-
i)
Maximum likelihood distances were calculated among haplotypes and used to construct a neighbor-joining tree using PAUP* 4.0b8 (PPC) (Swofford, 2000), incorporating a gamma-corrected HKY (Hasegawa-Kashino-Yano) model with parameters estimated from the dataset. Reliability of nodes defined by the phylogenetic tree was assessed using bootstrap re-sampling based on 500 replicates. A homologous sequence of a domestic pig (S. scrofa) was used as outgroup.
-
ii)
A minimum spanning network generated with the program TCS version 1.13 (Clement et al, 2000) was used to depict the phylogenetic, geographical and potential ancestor–descendant relationships among haplotypes. This cladogram construction procedure is specifically designed to estimate intraspecific gene trees where most of the haplotypes are present in the population.
Genetic variation and population structure
The nucleotide diversity index, π (Nei, 1987, equation 10.5), was used to estimate within-population genetic diversity. We analyzed population structure by analysis of molecular variance (AMOVA) as implemented in the program ARLEQUIN version 2.0 (Schneider et al, 2000). AMOVA divides total variance into additive components, that is, variation attributed to differences within populations, among populations within groups and among groups. Populations were grouped according to clades (or groups) identified in phylogenetic analysis. The statistical significance of the F statistics in AMOVA was assessed using 1000 random permutations.
Net sequence divergence between populations was used to estimate a population tree using the neighbor-joining algorithm implemented in PHYLIP version 3.5c (Felsenstein, 1993).
Analysis of microsatellite loci
Six highly polymorphic microsatellite loci originally described in the domestic pig (e.g. Rohrer et al, 1994,1996) were optimized and used in this study. These are SW607, S0289, SW1682, SW1301, SW403 and SW2419. All loci are dinucleotide repeats. SW607 and SW2419 lie on chromosome 6, but all other loci lie on different chromosomes on the porcine genome map. The PCR was carried out in a 10 μl reaction volume containing 10 ng of total genomic DNA, 2 mM MgCl2, 2 × PCR GOLD buffer (Boehringer Mannheim GMBH), 0.2 pmol of each of the dNTPs, 0.2 pmol of each primer and 0.4 U of AmpliTaq GOLD DNA polymerase (Boehringer Mannheim GMBH). PCR temperature profiles started with 94°C for 10 min of initial DNA denaturation and enzyme activation. This was followed by 28–34 cycles (28 for S0289; 30 for SW1682 and SW607; 32 for SW1301 and SW403; and 34 cycles for SW2419) of denaturation at 94°C for 30 s, annealing at 50–58°C (50°C for SW403; 52°C for SW607; 57°C for S0289 and 58°C for SW1882, SW1301 and SW2419) for 60 s and an extension at 72°C for 30 s. A final extension of 10 min followed all the reactions. All PCR reactions were run using dye-labeled primers (one primer in each primer set). The products were run on 4% acrylamide gels on ABI 377 (Perkin-Elmer) using ROX 500 as an internal standard.
Statistical analysis
An exact test based on a Markov chain algorithm was conducted to test deviations from Hardy–Weinberg proportions (Guo and Thompson, 1992) across loci in each population using the program GENEPOP version 3.1. (Raymond and Rousset, 1995). The Bonferroni correction for multiple comparisons was applied to the test. Genetic diversity within populations was measured as number of alleles per locus (A), observed heterozygosity per locus (Ho) and expected heterozygosity per locus (He) under Hardy–Weinberg expectations (Nei, 1987).
Microsatellite variation was analyzed in a hierarchical manner as was done for control region sequence data using the program ARLEQUIN version 2.0 (Schneider et al, 2000). The extent of genetic differentiation among populations and major groups was quantified using the ρ statistic with the program RSTCALC (Goodman, 1997). ρ is Goodman's (1997) analog of Slatkin's (1995) RST unbiased with regard to sample size. The statistical significance of ρ was assessed with 1000 permutations. Only locations with at least six individuals were included in the analysis of population structure (Table 1).
Results
Mitochondrial control region
Sequence characteristics and patterns: The d-loop showed moderate sequence variation, with 64 variable sites comprising 51 transitions, eight transversions and three deletions/insertions (Figure 2). Two sites showed both substitution categories. A total of 70 different haplotypes were observed, out of which 41 were scored only once. The most frequent haplotype was scored in 21 east African individuals; one from Maswa (Tanzania) and 20 from QE (Uganda). Sequences of these haplotypes have been submitted to GenBank (accession numbers AY253760–AY253829). Although sharing of haplotypes was observed between some populations within geographic regions, no such sharing was observed between regions (Figure 2). Within west Africa, 20 unique haplotypes were observed, 18 and 32 unique haplotypes were observed in southern and eastern Africa, respectively. Eight substitutions were exclusive to west Africa; six transitions, one transversion and one insertion/deletion.

Distribution of the 70 d-loop haplotypes observed in 181 warthogs from 24 localities. The vertical numbers indicate the position of polymorphic sites relative to haplotype GA 28. A dash (-) represents a deletion introduced to optimize alignment.
Phylogenetic relationships: Figure 3 shows the phylogenetic relationships of haplotypes within the common warthog. There are three major clades: western, southern and eastern African. Only one individual sampled in eastern Africa (TZ3 from Selous in Tanzania) clustered in the southern African clade and one individual from southern Africa (ZB7 from Zimbabwe) clustered in the eastern African clade. Each clade occurs within the range of a proposed subspecies.

Rooted neighbor-joining haplotype tree of the common warthog. A homologous sequence from the domestic pig (S. scrofa) was used for rooting. Numbers in parentheses are the numbers of individuals represented by that particular haplotype.
The minimum spanning network (Figure 4) supports the classification of the haplotypes into three main warthog clades representing western, southern and eastern African warthogs. Apart from two haplotypes, ZB7 sampled in Zimbabwe which clusters in the eastern African clade and TZ3 sampled in eastern Africa which clusters in the southern African, all others follow the geographical origin of individuals. The western African clade is separated from the southern African clade by 17 mutational steps and is also distinguished from the rest by eight fixed substitutions: six transitions, one transversion and one insertion/deletion. The sequence divergence between the western and eastern African clades is 6.6%, while that between the western and southern African clades is 4.3%. The eastern African clade is separated from the southern African clade by at least 10 mutational steps and a sequence divergence of 3.1%. All haplotypes in the western African clade were inferred to be derived from haplotype GA28 (sampled from Ghana in west Africa), while those in the eastern and southern African clades were derived from haplotype TZ15 (represented by individuals sampled from Tanzania and Uganda in eastern Africa) and NB1 (represented by individuals sampled from Namibia in southern Africa), respectively.

Minimum spanning network showing the phylogenetic relationship between the observed 70 mitochondrial control region haplotypes. Hatch marks along branches indicate number of nucleotide differences separating each haplotype in excess of one. Rectangles represent the inferred ancestral haplotypes. Haplotypes from the three different regions are indicated by different patterns.
Genetic diversity and population structure: Because of small sample sizes, only 12 localities, each with at least six samples and consisting of a total of 162 samples were used in the population study (Table 1). Nucleotide diversity in the total sample is 4.0%. Within populations, nucleotide diversity varies considerably, from as low as zero in QE and Tsavo to 2.1% in Zambia (Table 2). Nucleotide diversity in the three major lineages (hereafter referred to as subspecies) as revealed in phylogenetic analysis is 1.5% in both the western and eastern Africa lineages and 1.9% in the southern Africa lineage.
A hierarchical analysis of molecular variance of population structure reveals a highly significant subdivision between populations in the total sample (FST=0.853, P<0.001), between populations within each subspecies (FSC=0.524, P<0.001) and among the three subspecies (FCT=0.691, P<0.001). When pairwise comparisons of populations were made, all but one pair showed significant differentiation. The nondivergent population pair is between Zambia and Zimbabwe (southern Africa). The extent of differentiation among most population pairs was very high with approximately 70% of all pairwise comparisons showing FST values of more than 0.7 indicating limited exchange of breeding females between populations. An unrooted population tree (Figure 5a) based on net interpopulation distances groups the populations into three clusters that are concordant with their geographic origin and subspecific designations.

Population trees generated using (a) net interpopulation distance based on sequence data and (b) RST distances based on six microsatellite loci.
Microsatellite variation
Genotypic distribution and diversity: Allele size variation at six dinucleotide microsatellite loci was scored in a total of 143 warthog individuals from 11 localities in Africa (Table 3). Apart from one pair of loci located on the same chromosome, all other loci are located on different chromosomes on the porcine genome map, implying independent assortment for most loci used in this study.
Genotypic proportions at five loci in GA (SW1682, SW2419, S0289, SW607, S01301), two in QE (SW607 and SW24129) and one in MB (SW607) were significantly not in HW expectation after the Bonferroni correction. All significant P-values were due to an excess of homozygotes as indicated by the positive FIS values shown in Table 3. The statistics describing genotypic distribution and diversity for each population and locus are summarized in Table 3.
All loci analyzed in this study were highly polymorphic with total number of alleles per locus ranging from six at locus SW403 to 21 at locus S0289. A total of 100 different alleles were scored in all populations across all loci. The total number of different alleles scored in each population varies from 26 in MSA to 56 in GA (Table 4). The number of alleles per locus within populations range from two to 13 (Table 3). Overall levels of genetic diversity measured in terms of average expected heterozygosity were moderate to high for each population ranging from 0.59 in NB to 0.80 in GA (Table 3).
Population structure: As with the mtDNA data, the statistical analysis of population differentiation was performed for populations with at least six individuals. As KV individuals failed to amplify at most loci, only 11 populations with a total of 143 individuals were analyzed for microsatellite variation. A hierarchical analysis of molecular variance revealed highly significant subdivision between populations in the total sample (FST=0.199, P<0.001), among populations within each subspecies (FSC=0.134, P<0.001) and among the three subspecies (FCT=0.068, P<0.01). Unlike the mitochondrial loci where almost all population pairs were differentiated, only 35 out of 55 pairwise comparisons showed significant differentiation (P<0.05). However, despite the limited differentiation, all comparisons with the GA population resulted in highly significant RST values ranging from 0.162 (between GA and LR) to 0.300 (between GA and ZB) (P<0.001).
No significant population differentiation was observed among any population pairs from southern Africa (i.e. NB ZBA and ZB with all RST being negative), but within populations from eastern Africa, differentiation was observed at 13 out of 21 pairwise comparisons.
The allele frequency distribution at each locus in the different populations is shown in Table 4. A total of 21 population-specific alleles were observed over the six loci in the 11 populations. Of these alleles, 12 were scored in GA alone. At loci S0289 and SW403, rare alleles in the eastern and southern African population, respectively, become the most common alleles in GA. The eastern and southern Africa populations differ from each other by allele frequencies rather than by population-specific alleles.
A population tree based on RST is shown in Figure 5b. It differs from that produced using sequence data (Figure 5a) in that the most deviating population (GA) is connected to the populations from east Africa and not the populations from southern Africa. As in the sequence data, the grouping into western, eastern and southern Africa can be recognized.
Discussion
Intraspecific phylogeny of the common warthog
Our phylogenetic analyses (Figures 3 and 4) show that the common warthog we sampled comprises three divergent groups (southern, western and eastern African) with a sequence divergence ranging from 3.1 to 6.6%. This interpretation (which is provisional because data on the fourth subspecies was not included) is also supported by analysis of microsatellite data (Figure 5b). Except for the presence of tropical forests in the Congo Basin, there is no recent physical barrier between populations that can account for such sequence divergence. In the absence of a physical barrier, the geological events that might most plausibly explain this divergence are the climatic and habitat shifts of the Pleistocene. During adverse conditions, when dry climates and habitats expanded, populations of the warthog might have been isolated in three refugia in the west, east and south of the continent.
Our interpretation of genetic patterns also implies that:
-
i)
The desert warthog, which is the closest relative of the common warthog, is a product of earlier cycles. There is no comparable data on the desert warthog, but we predict that the two sister species form monophyletically reciprocal clades.
-
ii)
Climates in the Pleistocene must have been remarkably extreme to isolate warthogs considering that they are capable of surviving in harsh conditions. Indeed paleoclimatic evidence suggests that for much of this period, and earlier still, the African continent has been very dry, and today's climates are probably closer to the moist, warm end of the scale (Hamilton and Taylor, 1992).
Genetic diversity and population structure
The overall level of nucleotide diversity observed in the warthog is moderate (4.0%). This value is, however, misleading because it is due to a combination of three divergent subspecies. In each of these subspecies, nucleotide diversity is low. Within populations, levels of genetic diversity range from low to moderate at the mtDNA loci and medium to high at microsatellite loci (Tables 2 and 3). The mtDNA diversity estimates presented here cannot be directly compared to those of other pigs or their closest relatives, the peccaries, because equivalent data (on d-loop sequences) are unavailable for them. However, when compared to other large African mammals such as buffaloes (Simonsen et al, 1998), Grant's gazelle (Arctander et al, 1996), impala and greater kudu (Nersting and Arctander, 2001), warthogs show relatively low levels of mtDNA diversity. By contrast, variation at microsatellite loci observed in the common warthog is comparable to, or greater than that observed in some other large African mammals such as buffaloes (Simonsen et al, 1998), elephants (Nyakaana and Arctander, 1999) and waterbuck (Simonsen, 1997).
Analysis of genetic structure among warthog populations based on mtDNA sequence variation showed significant differentiation among all the localities. By contrast, microsatellite data showed weaker differentiation for many population pairs. The low genetic diversity but high differentiation at mitochondrial loci and high genetic diversity but low differentiation at nuclear loci can be interpreted in several ways:
-
i)
It could be evidence of male-biased dispersal among warthogs. This is, however, an unlikely explanation because both male and female warthogs are known to be strongly philopatric and there are no field studies indicating the contrary (e.g. Bradley, 1968; Cumming, 1975).
-
ii)
It could be a result of a more prevalent saturation of substitutions at microsatellite than at mitochondrial loci. Homoplasy has been reported among microsatellite alleles in a wide range of studies in the same population (e.g. Viad et al, 1998), in different populations (e.g. Blanquer-Maumont and Crouau-Roy, 1995; Orti et al, 1997) and between species (Blanquer-Maumont and Crouau-Roy, 1995). In the warthog, the failure to detect subdivision between geographically distant populations at microsatellite loci when such subdivision is detectable at the mitochondrial d-loop could suggest some saturation at the former loci.
-
iii)
An additional, and probably more likely, explanation for the different levels of differentiation at microsatellites and the mitochondrial d-loop lies in the fact that the microsatellites are highly variable. Hedrick (1999) pointed out that there is an upper limit for Wright's FST values. Instead of the theoretical upper limit of 1, highly variable loci have an upper bound of 1−H, where H is the average expected heterozygosity within populations. For the warthog, H=0.72 (Table 2), meaning that the upper bound is 0.28. We observed FST=0.20, meaning that the microsatellites are highly differentiated and approaching their upper limit.
Conclusion
Our results clearly indicate that we sampled three genetically distinct groups of the common warthog; one southern, one western and the other in eastern Africa. We also observed low genetic diversity but high differentiation at mitochondrial loci and high genetic diversity but low differentiation at nuclear loci in the warthog. These results have been interpreted in terms of the Pleistocene climatic cycles and high variability at microsatellite loci.
References
Amos W, Hoelzel AR (1991). Long-term preservation of whale skin for DNA analysis. In: Hoelzel AR (ed) Genetic Ecology of Whales and Dolphins, Report of the International Whaling Commission (Special Issue, 13). pp 99–104.
Arctander P, Kat PW, Aman AR, Siegismund RH (1996). Extreme genetic variation between populations of Grant's gazelle (Gazella grant) in Kenya. Heredity 76: 465–475.
Arctander P, Johansen C, Coutellec-Vreto M (1999). Phylogeography of three closely related African bovids (Tribe Alcelaphini). Mol Biol Evol 16: 1724–1739.
Arnason U, Gullberg A, Widegren B (1993). Cetacean mitochondrial control region: sequences of all extant baleen whales and two sperm whale species. Mol Biol Evol 10: 960–970.
Blanquer-Maumont A, Crouau-Roy B (1995). Polymorphism, monomorphism and sequences in conserved microsatellites in primate species. J Mol Evol 41: 492–497.
Bradley RM (1968). Some Aspects of the Ecology of the Warthog (Phacochoerus aethiopicus pallas) in the Nairobi National Park. MSc Thesis, University of Nairobi, Nairobi.
Clement M, Posada D, Crandall AK (2000). TCS: a computer program to estimate gene genealogies. Mol Ecol 9: 1657–1659.
Coope GR (1994). The response of insect faunas to glacial–interglacial climatic fluctuations. Phil Trans R Soc Lond B 344: 19–26.
Cumming DH (1975). A field study of the ecology and behaviour of Warthog. Museum memoirs of Salisbury.
deMenocal BP (1995). Plio-Pleistocene African climate. Science 270: 53–58.
Felsenstein J (1993). PHYLIP (Phylogeny Inference package) Version 3.52. Distributed by the author. Department of Genetics, University of Washington, Seatle.
Flagstad Ø, Syvertsen OP, Stenseth CN, Jakobsen SK (2001). Environmental change and rates of evolution: the phylogeographic pattern within the hartebeest complex as related to climatic variation. Proc R Soc Lond B 268: 667–677.
Gilbert SJ (1993). SeqApp 19 Biocomputing office, Biology Department, Indiana University of Bloomington.
Goodman SJ (1997). RSTCALC: a collection of computer programs for calculating unbiased estimates of genetic differentiation and gene-flow from microsatellite data and determining their significance. Mol Ecol 6: 881–886.
Grubb P (1993). The afrotropical suids (Phacochoerus, Hylochoerus and Potamochoerus). Taxonomy and description. In: WLR Oliver (ed) Pigs, Peccaries and Hippos. Status Survey and Conservation Plan, IUCN Gland: Switzerland.
Guo SW, Thompson EA (1992). Performing exact test for Hardy–Weinberg proportions for multiple alleles. Biometrics 48: 361–372.
Hamilton AC (1982). Environmental History of East Africa: a Study of the Quaternary. Academic Press: London.
Hamilton AC, Taylor D (1992). History of climate and forests in tropical Africa during the last 8 million years. In: Myers N (ed) The History of Tropical Forests, Kluwer: Dordrecht. pp 65–78.
Hedrick PW (1999). Highly variable loci and their interpretation in evolution and conservation. Evolution 53: 313–318.
Kim KI, Lee JH, Zhang YP, Lee SS, Gongora J, Moran C (2002). Phylogenetic relationships of Asian and European pig breeds determined by mitochondrial DNA D-loop sequence polymorphism. Anim Genet 33: 19–25.
Kingdon J (1989) East African Mammals. An Atlas of Evolution in Africa. The University of Chicago: Chicago. Vol.111B.
Meester J, Setzer WH (1971). The Mammals of Africa: An Identification Manual. Smithsonian Inst. Press.
Nei M (1987). Molecular Evolutionary Genetics. Columbia University Press: New York.
Nersting GL, Arctander P (2001). Phylogeography and conservation of impala and greater kudu. Mol Ecol 10: 711–719.
Nyakaana S, Arctander P (1999). Population genetic structure of the African elephant in Uganda based on variation at mitochondrial and nuclear loci: evidence for male-biased geneflow. Mol Ecol 8: 1105–1115.
Orti G, Pearse DE, Avise JC (1997). Phylogenetic assessment of length variation at microsatellite locus. Proc Natl Acad Sci USA 94: 10745–10749.
Paulo SO, Dias C, Bruford MW, Jordan WC, Nicholas RA (2001). The persistence of Pliocene populations through the Pleistocene climatic cycles: evidence from the phylogeography of Iberian lizard. Proc R Soc Lond B 268: 1625–1630.
Raymond M, Rousset F (1995). GENEPOP: population genetics software for exact tests and ecumenicism. J Hered 86: 284–249.
Rohrer GA, Alexander JL, Keele WJ, Smith PT, Beatie WC (1994). A microsatellite linkage map of the porcine genome. Genetics 136: 231–245.
Rohrer GA, Alexander LJ, Hu Z, Smith TPL, Keele JW, Bettie CW (1996). Cloning and characterization of 414 polymorphic porcine microsatellites. Anim Genet 27: 137–148.
Sanger F, Nicklen S, Coulson AR (1977). DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74: 5463–5467.
Sambrook J, Fritsch EF, Maniatis T (1989). Molecular Cloning: a Laboratory Manual. Coldspring Harbor Laboratory Press: Coldspring Harbor, NY.
Schneider S, Jean-Marc Kueffer, Roessli D, Excoffier L (2000). ARLEQUIN Ver.2.000: A software for population Genetic data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland.
Simonsen BT (1997). Population Structure and History of African Bovids. PhD Thesis, Department of Population Biology, Zoological Institute, University of Copenhagen.
Simonsen BT, Siegismund RH, Arctander P (1998). Population structure of African buffalo inferred from mtDNA sequences and microsatellite loci: high variation but low differentiation. Mol Ecol 7: 225–237.
Slatkin M (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462.
Swofford, DL (2000) PAUP*: Phylogenetic inference using parsimony (*and other methods), Version 4.0. Sinauer Assoc., Sunderland, MA.
Viad F, Frank P, Dubois MP, Estoup A, Jarne P (1998). Variation of microsatellite size homoplasy across electromorphs, loci and populations in three invertebrate species. J Mol Evol 47: 42–51.
White DT . (1995). Africa omnivors: global climatic change and Plio-Pleistocene hominids and suids. In: Vrba SE, Denton HG, Patridge CT, Burke HL (eds). Paleoclimate and Evolution with Emphasis on Human Origins, Yale University Press: New Haven, London.
White DT, Harris JM (1977). Suid evolution and correlation of African hominid localities. Science 198: 13–21.
Acknowledgements
This work was funded by the Danish International Development Agency (DANIDA) under the Wildlife Genetics Project, a collaborative project between the University of Copenhagen (Denmark) and Makerere University (Uganda). We are greatly indebted to the Wildlife authorities from Uganda, Kenya, Tanzania, Namibia and Zambia for their support. David Moyer from Tanzania and John Mason from Ghana are especially thanked for their sampling efforts.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Muwanika, V., Nyakaana, S., Siegismund, H. et al. Phylogeography and population structure of the common warthog (Phacochoerus africanus) inferred from variation in mitochondrial DNA sequences and microsatellite loci. Heredity 91, 361–372 (2003). https://doi.org/10.1038/sj.hdy.6800341
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800341
Keywords
This article is cited by
-
Phylogeographic Patterns in Africa and High Resolution Delineation of Genetic Clades in the Lion (Panthera leo)
Scientific Reports (2016)
-
Hidden diversity in Senegalese bats and associated findings in the systematics of the family Vespertilionidae
Frontiers in Zoology (2013)
-
Where is the game? Wild meat products authentication in South Africa: a case study
Investigative Genetics (2013)
-
Discordance Between Spatial Distributions of Y-Chromosomal and Mitochondrial Haplotypes in African Green Monkeys (Chlorocebus spp.): A Result of Introgressive Hybridization or Cryptic Diversity?
International Journal of Primatology (2013)
-
Phylogeographic analysis of nuclear and mtDNA supports subspecies designations in the ostrich (Struthio camelus)
Conservation Genetics (2011)
