
Abstract
The Saethre–Chotzen syndrome (SCS) is an autosomal dominant craniosynostosis syndrome with uni- or bilateral coronal synostosis and mild limb deformities. It is caused by loss-of-function mutations of the TWIST 1 gene. In an attempt to delineate functional features separating SCS from Muenke's syndrome, we screened patients presenting with coronal suture synostosis for mutations in the TWIST 1 gene, and for the Pro250Arg mutation in FGFR3. Within a total of 124 independent pedigrees, 39 (71 patients) were identified to carry 25 different mutations of TWIST 1 including 14 novel mutations, to which six whole gene deletions were added. The 71 patients were compared with 42 subjects from 24 pedigrees carrying the Pro250Arg mutation in FGFR3 and 65 subjects from 61 pedigrees without a detectable mutation. Classical SCS associated with a TWIST 1 mutation could be separated phenotypically from the Muenke phenotype on the basis of the following features: low-set frontal hairline, gross ptosis of eyelids, subnormal ear length, dilated parietal foramina, interdigital webbing, and hallux valgus or broad great toe with bifid distal phalanx. Functional differences were even more important: intracranial hypertension as a consequence of early progressive multisutural fusion was a significant problem in SCS only, while mental delay and sensorineural hearing loss were associated with the Muenke's syndrome. Contrary to previous reports, SCS patients with complete loss of one TWIST allele showed normal mental development.
Similar content being viewed by others


Introduction
Since its first description seven decades ago,1, 2 the Saethre–Chotzen syndrome (SCS) [MIM 101 400] has been well delineated as an autosomal dominant craniosynostosis syndrome of variable expression.3, 4 While early descriptions established the diagnosis on the basis of phenotype and pedigree studies, molecular genetics offers the opportunity to support the clinical diagnosis by mutation analysis of the TWIST 1 gene located on human chromosome 7p21.5, 6, 7 TWIST genes have been recognized as phylogenetically highly conserved genes encoding transcription factors, which probably influence other target genes involved in the induction of mesodermal tissues and cytokine expression through the NF-κB signal pathway.8, 9, 10, 11 There is also a connection to the FGF/FGFR cascade.12 Recently, an interaction between a C-terminal domain called TWIST box and the Runx2 pathway has been demonstrated to play an important role in osteoblast differentiation.13 Therefore, the TWIST box probably has to be added to the known important functional domains, that is, the DNA binding domain and the helix–loop–helix (HLH) motif.12 The SCS phenotype was found exclusively related to sequence changes within the first coding exon of TWIST 1, and haploinsufficiency of the gene was suggested as the underlying pathogenetic principle.7, 14 The clinician is faced with considerable phenotypic variation and some overlap with other syndromes, in particular with Muenke coronal synostosis [MIM 602 849], which is caused by a single recurrent mutation Pro250Arg in exon 7 of the FGFR3 gene.15, 16, 17 The differentiation between these closely related clinical entities requires considering the entire spectrum of phenotypic features, particularly those with therapeutic implications. The present investigation was aimed at the delineation of an entire spectrum of anomalies in TWIST-related SCS with emphasis on their functional significance, and attempts to establish criteria separating SCS from other entities of coronal craniosynostosis.
Methods of investigation
For the present study, we selected patients presenting with anterior plagiocephaly or brachycephaly due to uni- or bilateral coronal synostosis. If possible, first-degree relatives were examined as well. Subjects harboring a point mutation in the FGFR1 or FGFR2 gene18 or the mutation specific for the Crouzon phenotype with acanthosis nigricans (n=2) were not considered. Also, three patients affected with microscopic visible chromosomal deletions of the 7p21 region were excluded. The remaining 178 subjects were first classified according to their primary phenotypic diagnosis: SCS, Muenke's syndrome,16, 19 and undefined syndromes including nonsyndromic craniosynostosis (Table 1).
The SCS phenotype was defined according to the generally accepted criteria:20, 21 uni- or bicoronal synostosis, low-set frontal hairline, ptosis of eye lids, small palpebral fissures, prominent helical crura, digital webbing between second and third fingers, and a broad great toe with or without valgus deformity, or a bifid endphalanx on radiographs.22 The phenotypic diagnosis of Muenke's syndrome was based on the following features: uni- or bilateral coronal synostosis, brachydactyly, mental retardation, and sensorineural hearing deficit.16, 19
Clinical examination
Examination by at least one of the authors (WK, CS, GL, BP, HC) was supplemented by repeated photographic documentation. Syndromic features were defined according to standard criteria of the literature, using quantitative anthropometry for stature,23 head circumference, interorbital distance, and ear length.24 In addition, neurological, ophthalmic, and ENT evaluation as well as neurodevelopmental screening was performed by specialized team members. Plain radiographs of the skull and the cervical spine as well as CT scans and MR images were taken from most patients. Radiographs of the hands and feet were available in a subset of the group. Intracranial hypertension was defined by the presence of papilledema or by an intracranial baseline pressure exceeding 20 mmHg as registered by an epidural transducer during two night cycles.
Surgery and postoperative follow-up
Operative treatment was carried out in the majority of individuals and usually consisted of a forehead advancement procedure, supplemented by an extended cranial expansion in several patients suffering from overt intracranial hypertension. The age at first surgery varied from 2 months to 10 years, the median age being 15 months. Follow-up studies were performed in most subjects until late adolescence, and focused on recurrent intracranial hypertension, psychomotor development, and other functional sequelae. Long-term surveillance included repeated syndromological investigation, funduscopic examination at 3-month intervals, repeated radiographs of the skull, and a routine evaluation of midfacial growth, upper airway function, and hearing.
Mutation analysis
Blood samples were taken for diagnostic purposes after informed consent. DNA was extracted from white blood cells using standard procedures. Exon 5 of the FGFR1 gene and exons 8 and 10 of the FGFR2 gene were analysed according to protocols previously described.18 Exon 7 of the FGFR3 gene was amplified and digested with the restriction enzyme NciI to detect mutation Pro250Arg.15 Exon 1 of the TWIST 1 gene was amplified in two parts6 and sequenced directly on both strands (Beckman sequencer CEQ 8000).
Deletion mapping: In a first step, SCS-CA marker33 was routinely typed in patients without a mutation detectable during sequencing. If it was homozygous, a panel of polymorphic markers was typed consequently in the patient and his parents to detect loss of heterozygosity: D7S664 (F: AATTCTATCTTTCCAGGATTATCTG; R: GATCAGTGCTGGTATAATAGTAGGT; 55°C, 8% DMSO), D7S507 (F: TGCCCAATTCTCAGTGTT; R: CTACGTACATGGCTGCAA; 51°C, 4% formamide), D7S488 (F: TGTTAAGATGTTAGCTGTTGATCACT, R: TCCCTGTGCTTGAAACTGTCT; 58°C, 8% DMSO), SCS-CA33 (F: AGGAAAGCGGAGTCTCTG, R: GCCAAGGGTCCTTTGATT; 51°C, 8% DMSO), D7S2495 (F: ATATGGCTTCCCAGGGTTCT, R: CATCAATGAAATGAATGCTCAG; 57°C, 4% formamide), D7S2559 (F: AGCAAAGATCTGTGTAGCATCTG, R: CCTTCCGCTAGTTTCCTCAG; 59°C,1.7 mM MgCl2, 14% DMSO), D7S503 (F: ACTTGGAGTAATGGGAGCAG, R: GTCCCTGAAAACCTTTAATCAG; 55°C, 3% formamide), D7S654 (F: TTGCTGGTGATTTTCCAGGT, R: CCACTCACTCTGTGGCATTT; 55°C, 14% DMSO).
Reaction conditions were 100 ng DNA, 10 pmol of each primer, 2.5 μl 10 × buffer (200 mM Tris-HCl, pH 8.4, 500 mM KCl, 2 mM dNTP, 15 mM MgCl2), 0.2 μl 32P-dCTP, 1 U Taq polymerase, plus formamide or DMSO and H2O to get a final 25 μl reaction volume; 30 cycles (95°C/30 s, annealing 30 s, 72°C/30 s) were applied. Amplicons were run on 6% sequencing gels (1700 V), and gels were dried for autoradiography.
If a marker was not informative in a family, it was indicated by a question mark in Table 3; the smallest and largest possible size of a deletion was calculated from the distance of an informative marker relative to the TWIST 1 gene.
In family No. 38, a polymorphism in the polyglycine stretch of TWIST 1 gene was detected during sequencing (mother and daughter carried a 244insGGC) and subsequently used to show loss of heterozygosity.
Results
DNA analysis subdivided patients into four groups
Group 1A: Patients carrying missense mutations within the basic HLH (bHLH) domain of the TWIST 1 gene7, 14 or stop/frameshift mutations outside the bHLH domain, or large deletions spanning the entire gene.
Group 1B: Patients with sequence variations of the TWIST 1 gene, which did not meet the criteria of group 1A.
Group 2: Patients with the Pro250Arg substitution in the FGFR3 protein.15, 16
Group 3: Patients without detectable mutations within FGFR1–3 and TWIST 1. As mentioned above, patients with mutations in FGFR1 and FGFR2 had been excluded before.
A detailed analysis of mutations and re-assessment of the corresponding phenotypes revealed the following findings.
Group 1A: Within this group, a total of 24 different mutations were identified located at various sites of the HLH domain (n=14), the DNA binding region (n=3), and the N-terminal part of the TWIST 1 gene (n=7) (Table 2). We identified 13 novel mutations since the surveys of Gripp et al,7 Elanko et al,25 and Cai et al.26 In addition, six deletions of the entire gene region were detected (Tables 2 and 3). The 70 individuals of this group represented 38 independent pedigrees, and were divided up into 54 familial and 16 sporadic cases. In seven of the latter, the de novo origin could not be confirmed, as at least one of the parents was not available for molecular testing. There was one family carrying two separate point mutations on the same allele (pedigree No. 20; Table 2).
Group 1B: This group consisted of five sequence variants, but only one of them was associated with a clearcut SCS phenotype. This female patient (pedigree No. 36; Table 2) carried a novel missense mutation in a motif called TWIST box containing codons 183–202 of the TWIST 1 gene.13 She presented with typical clinical signs of SCS including plagiocephaly, progressive sutural fusion, intracranial hypertension, enlarged parietal foramina, low frontal hairline, blepharophimosis, prominent helical crura, and digital webbing, while ptosis and bifid great toe were absent. In addition to forehead advancement, at the age of 13 months she required cardiac surgery for a large ventricular septal defect. Until age 14 years, she had experienced a few epileptic seizures, and her mental performance was considered borderline although she attended a regular school.
(GGC)5GCG(GGC)5 repeat variations within TWIST 1 (c. 244–276)25 were incidentally detected during genetic screening. Three in-frame deletions within the 5′-DNA binding domain were phenotypically associated with isolated oxycephaly (pedigree No. 7), with normal phenotype in a mother of a patient with isolated trigonocephaly (pedigree No. 8), and with a Crouzon genotype due to an Arg768Gly substitution in exon 17 of FGFR2 in male twins and their non-affected mother (pedigree No. 9). A boy with isolated scaphocephaly and his healthy mother (pedigree No. 37) had a point mutation at the very 3′-edge of the TWIST box. These four cases were considered to represent non-disease-causing polymorphisms.
Inter- and intrafamilial phenotypic expression of TWIST 1-related SCS was highly variable (Table 2): While 62 of all 71 patients presented with typical features of SCS, nine cases of very mild expression had not been appreciated through inspection alone, and three of them only by means of pedigree analysis. Conversely, no subject presented with a phenotype suggestive of Crouzon's, Pfeiffer's, or Apert's syndrome.
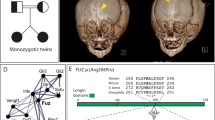
In addition to the commonly reported phenotypic criteria, we noted some features that contribute to a comprehensive spectrum of clinical signs (Table 4). These additional features included (1) a wide gap at the sagittal suture during infancy similar to that known from the Apert's syndrome, which eventually closed to result in complete fusion before the age of four (Figure 1a and b), and (2) multisutural fusion including the lambdoid suture as detected in 53% of subjects (Figure 1c). This latter finding corresponded to a surprisingly high rate of intracranial hypertension, which was proven in 35% of patients at a median age of 30 months. Papilledema was the most common sign, and was detected in 19 patients while epidural pressure monitoring was diagnostic in five others. Elevated intracranial pressure was successfully controlled by a variety of cranial expansion procedures, although 18% of surgically treated patients required a second procedure because of recurrent intracranial hypertension. Five patients not treated in time eventually developed significant visual loss due to irreversible optic nerve damage. One of these patients turned blind. Other causes of elevated intracranial pressure such as hydrocephalus were not encountered.

(a–d) Radiographs of the proposita of pedigree No. 20. (a, b) At birth, bilateral coronal synostosis and a large calvarial midline defect are present. (c) At 18 months, a Wormian bone fills the midline defect, and an ordinary sagittal suture is missing. Abnormal appearance of the lambdoid suture is shown. (d) At 36 months, all sutures have fused, and the parietal foramina are enlarged. Bilateral papilledema has developed.
A total of 37% of subjects presented with a head circumference two standard deviations below average. Stature and mental development were usually within normal limits. Even those six subjects with complete loss of one TWIST allele were mentally normal. There was no instance of sensorineural hearing deficit while conductive hearing deficit due to recurrent middle ear effusions was a common and significant problem.
In order to arrive at a clinical diagnosis of SCS, we found the following anomalies to be most helpful: ear length below two standard deviations of the norm,24 enlarged parietal foramina (i.e. >5 mm diameter) (Figure 1d), interdigital webbing, and a broad hallux or hallux valgus with a bifid or duplicated distal phalanx on radiographs (Table 5) (Figure 2). A total of 87% of evaluable subjects with SCS presented with at least one of these signs, and 53% exhibited two or more. None of these signs was observed in either Muenke's syndrome or nonsyndromic coronal synostosis except one single patient without a detectable mutation, who presented with bifid hallux (see below). In addition, ptosis of the eyelids severe enough to warrant surgical treatment was noted in 13 patients, whereas patients with Muenke's syndrome had mild ptosis at most. A low-set frontal hairline was considered typical for SCS patients, provided that ethnic variation was taken into account.

Radiograph of the right forefoot of the proposita of pedigree No. 21 at the age of 12, showing notching of the first terminal phalanx.
Group 2 (Pro250Arg in FGFR3): A total of 42 subjects carried the Pro250Arg exchange in the gene product of FGFR3. They represented 13 sporadic and 29 familial cases within 24 independent pedigrees. Clinical features consisted of uni- or bilateral coronal synostosis, a bulging forehead during infancy, marked temporal bulging, hypertelorism in nearly half of the patients, downslanting palpebral fissures, mild ptosis of eyelids, mild midface hypoplasia, and high arched palate. Additional features included brachydactyly of hands and feet, sensorineural hearing deficit, and also conductive hearing deficit due to recurrent middle ear effusions, and mental retardation (Table 5). As with the SCS patients, the phenotypic expression of Muenke patients varied considerably, ranging from a striking deformity to an essentially normal appearance even within the same pedigree.
Few affected subjects of this group had been identified by means of classical syndromology. Confusion with TWIST 1-related SCS frequently occurred prior to the detection of this particular mutation of FGFR3, and was due to an overlap of some phenotypic features such as hypertelorism, mild eyelid ptosis, anti-mongoloid slant of palpebral fissures, and brachydactyly. In contrast to TWIST 1-related SCS, clinically significant ptosis was not encountered in any of the Muenke patients. The major differences between the two entities relate to intracranial pressure and brain function. Only two Muenke's syndrome patients presented with an equivocal finding of increased dural tension during surgery, while no instance of papilledema or optic nerve atrophy was encountered. Progressive craniosynostosis involving at least the sagittal suture was found in a considerable number of patients. The lambdoid suture was affected in three of the patients as well. However, in Muenke's syndrome patients, these abnormalities appeared at a later age (median age 7 years; range 4–16 years) than in SCS (median age 4 years; range 8 months to 12 years). Mental retardation occurred in roughly one-third of evaluable Muenke's syndrome patients, and generally was of mild to moderate degree in all but two who had severe deficits. Sensorineural hearing deficit was an exclusive feature of Muenke's syndrome in the present series (Table 5).
Group 3 (coronal synostosis without identified mutation): In 61 independent pedigrees (65 individuals), molecular screening failed to detect any mutation at the hot spots of the investigated genes, that is, FGFR1 and FGFR2, TWIST 1, and the Pro250Arg mutation in FGFR3. Since associated anomalies were actually absent in most cases, classification as an isolated nonsyndromic craniosynostosis phenotype appeared justified in those patients (Table 1). Nevertheless, as expected from the limitations of our genetic screening methods, this group included a number of patients who met the criteria of syndromic, yet unclassified craniosynostosis. Three patients exhibited some features of SCS phenotype including mild ptosis (n=2), brachydactyly (n=2), and duplicated distal phalanx of the great toe (n=1). Another patient was misdiagnosed as Muenke's syndrome because of associated mental retardation, and nine patients were affected with a variety of other unclassified syndromes including two cases with a Pfeiffer's syndrome phenotype. Also, there were four instances of familial isolated coronal synostosis. Intracranial hypertension due to craniostenosis was found in three patients with a phenotype of isolated synostosis, one of them developing papilledema. Craniosynostosis proceeded to involve the sagittal suture, and sometimes the lambdoid suture as well, in 36% of these patients at a median age of 87 months. Inner ear function was normal in all of these patients, and 11% showed various degrees of mental retardation.
Discussion
A multitude of different TWIST 1 gene mutations is associated with the SCS phenotype. All point mutations are located within the first coding exon, and most of these affect the DNA binding and the HLH domains, which until recently have been considered the main functional domains.7 Extending the surveys of Gripp et al,7 Elanko et al,25 and Cai et al,26 we add 14 novel point mutations and six large deletions including the entire TWIST 1 gene (Table 2) that are associated with the SCS phenotype. Since different mutations like missense mutations, nonsense mutations, insertions, and whole-gene deletions result in similar phenotypes, haploinsufficiency is the likely disease-causing mechanism.6, 7 In fact, many mutations associated with SCS abolish normal translation of the coding region. This is in contrast to the Muenke's syndrome, where the only recurrent mutation in the FGFR3 gene, Pro250Arg, results in a gain of function.27
The novel missense mutation Phe187Leu in the 3′-HLH region (pedigree 36) is located within a highly preserved motif called TWIST box,13 the binding region for the transcription factor Runx2 at which the anti-osteogenic function of the TWIST genes is exerted. To our knowledge, this is the second reported mutation within this domain causing a classic SCS phenotype.7 Remarkably, the Ser201Tyr exchange (pedigree 37) at the 3′-edge of the TWIST box in our pedigree 37 is obviously a rare polymorphism, which we did not find in more than 500 sequenced DNA samples. This exchange occurred in a boy with simple scaphocephaly and in his healthy mother without craniosynostosis.
Several sequence variations within the N-terminal part of the TWIST 1 gene (in front of the DNA binding domain) have been recognized as non-disease-causing polymorphisms. Elanko et al25 have provided arguments in favor of such polymorphisms within a polyglycine stretch beginning at cDNA position 244, which fit to our pedigrees with in-frame deletions affecting this gene region. The specificity of the genotype–phenotype correlation in SCS has been challenged by cases presenting with features of Crouzon, Baller–Gerold, or Robinow–Sorauf phenotypes carrying mutations in the TWIST 1 gene.26, 28, 29, 30
In the present series, TWIST gene mutations were exclusively associated with a typical SCS phenotype, albeit of quite variable expression. Not a single patient presented with the classical SCS phenotype without carrying a TWIST 1 gene mutation. Because of the phenotypic overlap, some authors have suggested to combine SCS and Muenke's syndrome into a single entity presenting with a broad phenotypic spectrum.17 In view of the striking differences in terms of function and morbidity, our data do not support this contention. In contrast to the Muenke phenotype, the SCS phenotype is characterized by a high risk of intracranial hypertension and a low risk of concurrent mental retardation. Patients carrying TWIST 1 mutations obviously bear a considerable risk for optic nerve damage from increased intracranial pressure,20 while no instance of papilledema was observed in the Muenke's syndrome.
Intracranial hypertension in SCS, obviously related to progressive synostosis, has not widely been appreciated as a prominent feature even though elevated intracranial pressure had been mentioned in the early descriptions of the SCS.2, 20, 31 The proportion of cases with elevated intracranial pressure in the present series is comparable to the situation in Apert patients.32
With respect to mental performance, carriers of TWIST mutations in the present series did not differ from the average population.20 Contrary to other authors,33, 34 we found that even deletions of the entire TWIST gene did not adversely affect cognitive function in our SCS patients. Compared to three deletion cases reported by Johnson et al,33 the size of the deletions in our patients appears to be smaller, particularly in the 3′-direction (Table 3), but further analysis is necessary. In the Muenke's syndrome, mental impairment ranging from mild to severe has been reported a common feature.16, 19 Sensorineural hearing deficit was exclusively noted in our patients with Muenke's syndrome, and has only rarely been reported in SCS.35 In summary, our study suggests that with regard to clinical management and genetic counselling, patients with putative SCS or Muenke phenotypes would benefit more from splitting than from lumping.
Our own patient series suggests that the prevalence of SCS, Muenke's syndrome, and isolated coronal synostosis may not differ very much from each other. FGFR-associated craniosynostosis has a high de novo mutation rate of about 50% with exclusive paternal origin,36 whereas only a third of all TWIST-associated SCS cases seem to be caused by de novo mutations.
Considering the wide range of phenotypic variability, the clinical suspicion of SCS should be confirmed by molecular analysis. However, in clinical practice, a preliminary diagnosis is possible on the basis of the most specific features and pedigree analysis. Once the diagnosis has been established, particular attention should be paid to intracranial hypertension, recurrent middle ear effusions, impending amblyopia resulting from ptosis, tear duct stenosis, and airway obstruction. Vertebral fusions and scoliosis might be present as well. Long-term surveillance should continue until adolescence and should include ophthalmoscopy in 3-month intervals even after successful skull surgery. Annual skull X-rays should be taken in order to detect progressive sutural fusion, which is associated with an increased risk of intracranial hypertension. In doubtful cases, intracranial pressure monitoring is recommended.
References
Saethre H : Ein Beitrag zum Turmschädelproblem. (Pathogenese, Erblichkeit und Symptomatologie.). Dtsch Z Nervenheilk 1931; 117: 533–555.
Chotzen F : Eine eigenartige familiäre Entwicklungsstörung. (Akrocephalosyndaktylie, Dystosis craniofacialis und Hypertelorismus.). Monatsschr Kinderheilk 1933; 55: 97–122.
Friedman JM, Hanson JW, Graham B, Smith DW : Saethre–Chotzen syndrome: a broad and variable pattern of skeletal malformations. J Pediatr 1977; 91: 929–933.
Reardon W, Winter RM : Saethre–Chotzen syndrome. J Med Genet 1994; 31: 393–396.
Howard TD, Paznekas WA, Green ED et al: Mutations in TWIST, a basic helix–loop–helix transcription factor, in Saethre–Chotzen syndrome. Nat Genet 1997; 15: 36–41.
El Ghouzzi V, Le Merrer M, Perrin-Schmitt F et al: Mutations of the TWIST gene in the Saethre–Chotzen syndrome. Nat Genet 1997; 15: 42–46.
Gripp KW, Zackai EH, Stolle CA : Mutations in the human TWIST gene. Hum Mutat 2000; 15: 150–155.
Yousfi M, Lasmoles F, Lomri A, Delannoy P, Marie PJ : Increased bone formation and decreased osteocalcin expression induced by reduced Twist dosage in Saethre–Chotzen syndrome. J Clin Invest 2001; 107: 1153–1161.
Jabs EW : A TWIST in the fate of human osteoblasts identifies signaling molecules involved in skull development. J Clin Invest 2001; 107: 1075–1077.
Wilkie AO, Morris-Kay GM : Genetics of craniofacial development and malformation. Nat Rev Genet 2001; 2: 458–468.
Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN : TWIST regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell 2003; 112: 169–180.
Castanon I, Bayliss MK : A Twist in fate: evolutionary comparison of Twist structure and function. Gene 2002; 287: 11–22.
Bialek P, Kern B, Yang X et al: A twist code determines the onset of osteoblast differentiation. Dev Cell 2004; 6: 423–435.
El Ghouzzi V, Lajeunie E, Le Merrer M et al: Mutations within or upstream of the basic helix–loop–helix domain of the TWIST gene are specific to the Saethre–Chotzen syndrome. Eur J Hum Genet 1999; 7: 27–33.
Bellus GA, Gaudenz K, Zackai EH : Identical mutations in three different fibroblast growth factor receptor genes in autosomal dominant craniosynostosis syndromes. Nat Genet 1996; 14: 174–176.
Muenke M, Gripp KW, McDonald-McGinn DM : A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet 1997; 60: 555–564.
Paznekas WA, Cunningham ML, Howard TD et al: Genetic heterogeneity of Saethre–Chotzen syndrome, due to TWIST and FGFR mutations. Am J Hum Genet 1998; 62: 1370–1380.
Kress W, Collmann H, Büsse M, Halliger-Keller B, Mueller CR : Clustering of FGFR2 gene mutations in patients with Pfeiffer and Crouzon syndromes (FGFR2-associated craniosynostosis). Cytogenet Cell Genet 2000; 91: 134–137.
Renier D, El Ghouzzi V, Bonaventure J, Le Merrer M, Lajeunie E : Fibroblast growth factor receptor 3 mutation in nonsyndromic coronal synostosis: clinical spectrum, prevalence, and surgical outcome. J Neurosurg 2000; 92: 631–636.
Pantke OA, Cohen MM, Witkop CJ : The Saethre–Chotzen syndrome. Birth Defects 1975; 11: 190–225.
Cohen MM : Saethre–Chotzen syndrome; in Cohen MM, MacLean RE (eds): Craniosynostosis – diagnosis, evaluation, and mangement. New York, 2000, chapter 28, pp 374–376.
Trusen A, Beissert M, Collmann H, Darge K : The pattern of skeletal anomalies in the cervical spine, hands and feet in patients with Saethre–Chotzen syndrome and Muenke-type mutation. Pediatr Radiol 2003; 33: 168–172.
Prader A, Largo RH, Molinari L, Issler C : Physical growth of Swiss children from birth to 20 years of age. Helvet Paediatr Acta Suppl 1989; 52: 1–125.
Hall JG, Froster-Iskenius UG, Allanson JE : Handbook of normal physical measurements. Oxford: Oxford University Press, 1989.
Elanko N, Sibbring JS, Metcalfe KA : A survey of TWIST for mutations in craniosynostosis reveals a variable length polyglycine tract in asymptomatic individuals. Hum Mutat 2001; 18: 535–541.
Cai J, Shoo BA, Sorauf T, Jabs EW : A novel mutation in the TWIST gene, implicated in Saethre–Chotzen syndrome, is found in the original case of Robinow–Sorauf syndrome. Clin Genet 2003; 64: 79–82.
Ibrahimi OA, Zhang F, Eliseenkova AV, Linhardt RJ, Mohammadi M : Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum Mol Genet 2004; 13: 69–78.
Gripp KW, Stolle CA, Celle L et al: TWIST gene mutation in a patient with radial aplasia and craniosynostosis: further evidence for heterogeneity of Baller–Gerold syndrome. Am J Med Genet 1999; 82: 170–176.
Kunz J, Hudler M, Fritz B : Identification of a frameshift mutation in the gene TWIST in a family affected with Robinow–Sorauf syndrome (letter). J Med Genet 1999; 36: 650–652.
Seto ML, Lee SJ, Sze RW, Cunningham ML : Another TWIST on Baller–Gerold syndrome. Am J Med Genet 2001; 104: 323–330.
Kopysz Z, Statiska M, Rysko I, Kulzyk B : The Saethre–Chotzen syndrome with partial bifid of the distal phalanges of the great toes. Hum Genet 1980; 56: 195–204.
Renier D, Arnaud E, Cinalli G et al: Prognosis for mental function in Apert's syndrome. J Neurosurg 1996; 85: 66–72.
Johnson D, Horsley SW, Moloney DM et al: A comprehensive screen for TWIST mutations in patients with craniosynostosis identifies a new microdeletion syndrome of chromosome band 7p21.1. Am J Hum Genet 1998; 63: 1282–1293.
Cai J, Goodman BK, Patel AS et al: Increased risk for developmental delay in Saethre–Chotzen syndrome is associated with TWIST deletions: an improved strategy for TWIST mutation screning. Hum Genet 2003; 114: 68–76.
Lee S, Seto M, Sie K, Cunningham M : A child with Saethre–Chotzen syndrome, sensorineural hearing loss, and a TWIST mutation. Cleft Palate Craniofac J 2002; 39: 110–114.
Goriely A, McVean GAT, Röjmyr M, Ingemarsson B, Wilkie AOM : Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science 2003; 301: 643–646.
den Dunnen JT, Antonarakis SE : Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000; 20: 7–12.
Acknowledgements
We thank all patients and their families as well as the ‘Elterninitiative Apert-Syndrom’ for supporting this study, and Birgit Halliger-Keller and Olga Kari for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kress, W., Schropp, C., Lieb, G. et al. Saethre–Chotzen syndrome caused by TWIST 1 gene mutations: functional differentiation from Muenke coronal synostosis syndrome. Eur J Hum Genet 14, 39–48 (2006). https://doi.org/10.1038/sj.ejhg.5201507
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201507
Keywords
This article is cited by
-
Deviating dental arch morphology in mild coronal craniosynostosis syndromes
Clinical Oral Investigations (2019)
-
Destabilization of the TWIST1/E12 complex dimerization following the R154P point-mutation of TWIST1: an in silico approach
BMC Structural Biology (2018)
-
RETRACTED ARTICLE: Fetal methotrexate syndrome and Antley–Bixler syndrome should not be confused
Pediatric Radiology (2018)
-
Craniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling
Journal of Applied Genetics (2018)
