
Abstract
Adaptation to endoplasmic reticulum (ER) stress relies on activation of the unfolded protein response (UPR) and induction of autophagy. Indeed, cells die if ER stress is not countered by the UPR. Here we show in U937 cells that the ER stressors tunicamycin and thapsigargin cause increased expression of c-Jun N-terminal kinase 2 (JNK2), which allows regulation of the UPR, whose silencing or pharmacological inhibition delays BiP (immunoglobulin heavy-chain binding protein) upregulation, and causes earlier and greater expression of CCAAT/enhancer-binding protein-homologous protein (CHOP). Furthermore, we show that pharmacological inhibition or silencing of JNK2 causes accumulation of both p62 and the acidic compartment, caspase 3 activation and apoptosis. Our results reveal that JNK2 prevents accumulation of the acidic compartment in U937 cells undergoing autophagic flux and, by this mechanism, it keeps stressed cells alive. Our findings highlight a potential role for JNK2 in tumor cell survival, senescence and neurodegenerative diseases, in which ER stress, autophagy and lysosome activity are known to interplay.
Similar content being viewed by others


Main
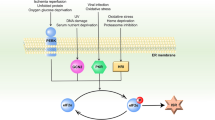
The endoplasmic reticulum (ER) has two vital roles in the cell – it is a major intracellular calcium store and it enhances post-translational modifications of newly synthesized proteins destined for secretion, cell surface expression or organelle biogenesis. The post-translational protein modifications that normally occur in the ER include folding, glycosylation, disulfide bond formation and protein trafficking between ER and the Golgi apparatus. Disturbances in ER calcium homeostasis and protein processing cause the accumulation of misfolded or unfolded proteins in the ER and initiate the unfolded protein response (UPR).1, 2 Transcription of UPR target genes, such as the ER molecular chaperone BiP/GRP78 (immunoglobulin heavy-chain binding protein/78 kDa glucose-regulated protein), which has an antiapoptotic and cytoprotective role in early embryogenesis, oncogenesis, neurodegenerative diseases and atherosclerosis, is promoted by inositol-requiring kinase 1 (IRE1) and ATF6.3, 4 More severe ER stress has been linked with cell death through the activation of the ER-associated caspase 12 and increased expression of CCAAT/enhancer-binding protein-homologous protein (CHOP)/GADD153, a transcription factor that sensitizes the cells to apoptosis.5, 6 As a survival mechanism, the UPR may upregulate the autophagy machinery to clear the ER of the accumulation of misfolded proteins that cannot be degraded by the proteasome,7, 8, 9, 10 using the ER itself to provide the membrane needed for autophagosome formation. Autophagy therefore appears to be an efficient mechanism for eliminating unfolded proteins, thereby contributing to ER homeostasis.10, 11
Nevertheless, excess stimulation of autophagy, as in the case of the UPR, can activate the cell death machinery,12, 13 making it likely that UPR and autophagy are interlinked, sharing a cytoprotective role under basal conditions or metabolic stress, and taking on a cytocidial function after acute cell damage. In this multistep and multifunctional crosstalk among organelles, the lysosomes appear highly dynamic, as they receive materials from trans-Golgi, endocytic and autophagic pathways.14, 15 They contain more than 50 hydrolases, which consent reuse of the breakdown products of the major cellular macromolecules. Lysosomes may contribute to angiogenesis, tumor growth and metastasis formation.16 Indeed, enhanced expression and secretion of lysosomal cysteine cathepsins has been observed in tumor cells. Leakage of cathepsins into the cytosol can also trigger necrotic and apoptotic cell death,17, 18 and consequently the maintenance of lysosomal membrane integrity is of utmost importance for cell survival. It is therefore axiomatic that the molecular pathways linking ER stress with either survival or death mechanisms need to be strictly regulated at multiple levels.
Previous studies indicated that c-Jun N-terminal kinase (JNK) activation may be linked to ER stress by IRE1,19 and that autophagy induction after ER stress relies on the IRE1-JNK pathway.7, 8 Indeed, JNK is known to regulate starvation-induced autophagy through Bcl-2 phosphorylation, thereby enabling cell survival.20 It also has a key role in autophagic cell death by mediating beclin 1 expression.21 Furthermore, it has been shown that ER-dependent cell fate may be regulated by the activation of JNK/ERK.22 JNK is a set of enzymes activated by dual phosphorylation by means of MAPK (mitogen-activated protein kinase) kinases MKK4 and MKK7 in response to a variety of stress signals, including UV irradiation, chemotherapy damage, osmotic stress, hypoxia/anoxia and hyperthermia.
Whether JNK has a prosurvival or a prodeath role is still, however, a controversial issue.20, 21, 22, 23, 24 This controversy may be explained by the selective interaction of JNK protein kinase isoforms with different transcription factors.25, 26 Indeed, there are numerous JNK substrates, which are known to have pleiotropic biological roles. To produce the 10 JNK isoforms, the three JNK genes are spliced differently, either in the center (a versus b) or at the COOH terminus (1 versus 2). Alternative splicing at the COOH terminus of the three JNK genes yields proteins that are either 46 or 54 kDa in size. Temporal regulation of JNK may also be a critical determinant of cellular responses: JNK1 transient activation, for example, promotes cell survival, whereas prolonged activation is known to mediate apoptosis.27
To clarify this issue, we set out to investigate whether moderate ER stress, caused by opportune doses of tunicamycin (TN), an N-glycosylation inhibitor, or thapsigargin (TG), which inhibits ER Ca2+ release, can in fact promote cell survival. Interestingly, by this means we identified a JNK2-dependent mechanism that appears to counter apoptotic cell death.
Results
ER stressors and cell survival
TN and TG are ER stressors that cause cell death in a dose-dependent manner: 24 h treatments with high concentrations of these drugs were cytotoxic for a large proportion of U937 cells, while lower concentrations were less effective, suggesting the activation of a survival pathway (Figures 1a and b). In particular, TN 1 μM caused a slight decrease in Trypan blue-excluding cells (85±3%) in comparison with untreated cultures (95±2), in addition to the appearance of 19±3% of sub-G1 events. Similarly, TG (200 nM) caused a fall in cell viability (84±4%) and the appearance of sub-G1 events (16±4%). Sub-G1 events were studied by cytofluorimetry of cell cycle phases of cells fixed and stained with propidium iodide (PI): hypodiploid DNA events are easily discernable from the narrow peak of cells with diploid DNA content, and are considered to be indicative of apoptotic nuclei.28 Furthermore, analysis of events in the different cell cycle phases showed that TN (1 μM) and TG (200 nM) caused a decrease in S and G2M phases, while the percentage of G1 events was apparently unchanged (Figure 1c). Cell counts indicate that neither treatment allowed cell growth (not shown). These results suggest that TN or TG, at such concentrations, cause activation of a survival pathway in most U937 cells, increasing the time spent in the G1 cell cycle phase. In this phase, cells may try to escape from the stress before it can cause serious damage to their functions. To investigate these survival pathway(s), the experiments were performed using TN (1 μM) and TG (200 nM), and viability parameters were investigated after 24 h.

Cell survival depends on the dose of ER stressors. U937 cells were exposed or not to different concentrations of TN (a) or TG (b) for 24 h. Cells were counted in a hemocytometer as excluding and not excluding Trypan blue. Viability was obtained by calculating live (Trypan blue-excluding) cells as a percentage of all counted cells. A portion of cells were fixed and stained with propidium iodide; sub-G1 events in the cell cycle were evaluated by cytofluorimetry. The reported values represent the means and the error bars the S.D. of the percentage of live cells (Trypan blue-excluding) or sub-G1 events of 10 independent experiments. Assessment of cell survival was investigated and statistically significant differences (P<0.05) were found between the data obtained using TN 1 μM or TG 200 nM and the two highest concentrations of each drug used. (c) The cell cycle profiles of one representative experiment out of 10 performed on U937 cells untreated (upper panel) or treated with TN (1 μM) (middle panel) or with TG (200 nM) (lower panel) for 24 h. The percentage of events in the different cell cycle phases quantified in the reported experiment are indicated in each panel
ER stressors induce activation of JNK2
JNK has been ascribed a role in ER-mediated survival.7, 8, 20, 22 To investigate its involvement in the previously detected survival pathway activated by TN or TG, we performed time-kinetics studies in which phosphorylated JNK and then total JNK were analyzed by western blot with specific antibodies. A band of 46 kDa of phospho-JNK proteins was detected in the lysate of untreated U937 cells, together with a much smaller band of 54 kDa proteins (Figure 2a). Whereas the former band remained unchanged upon ER stress, the amount of 54 kDa phospho-JNK proteins increased after 3 h and declined thereafter. When probed with antibodies against total JNK, in comparison with that observed in the lysate of untreated cells, the 46 kDa band showed no change at the investigated time points of ER stress, whereas the 54 kDa band increased after 3 h, and then declined after 6 h (Figure 2a). Similar results were obtained with TG (not shown). Thus, ER stressors upregulate and activate 54 kDa JNK proteins in U937 cells.

TN and TG induce JNK2 migration as a 54 kDa band. (a) JNK was detected by western blot in the lysates of U937 cells either exposed to TN (1 μM) or not for the time indicated. Blotted proteins were probed with anti-phospho-JNK and then anti-JNK-antibodies, each followed by peroxidase-conjugated secondary antibody. The level of β-actin is shown at the bottom as a loading control. (b) Western blot analysis of JNK, detected by antibody recognizing JNK, in the lysates of U937 cells treated with anisomycin (5 μM × 45 min) or transfected with equal amounts of JNK1- or JNK2-siRNA or scr-siRNA for 24 h and exposed to TN (1 μM) or TG (200 nM) for a further 3 h. Western blot analysis of β-actin is shown at the bottom as a loading control and also to confirm the specificity of the transfected siRNA. Representative blots are shown. Densitometric quantification of the bands is shown at the bottom of the relevant lines as the ratio of p54 in each line to the p54 value observed in the lysate of scr-siRNA-treated cells
The 46 or 54 kDa proteins detected may be produced by alternative splicing at the COOH terminus of JNK genes. To determine the relative contribution of JNK1 and JNK2 proteins to each of these bands, the cells were transfected with scrambled-small interfering RNA (scr-siRNA) or siRNA specific for either JNK1 or JNK2 for 24 h, followed by 3 h treatment with TN or TG. JNK expression was analyzed by western blot. In comparison with scr-siRNA, JNK2-targeting siRNA did not modify the 46 kDa band, but brought about an effective reduction (85–95%) in the 54 kDa band. In contrast, JNK1-targeting siRNA modified neither the 54 kDa band nor the 46 kDa band (Figure 2b). The electrophoretic mobilities of the two JNK bands corresponded to those observed in U937 cells treated with anisomycin (Figure 2b). These results were obtained with both TN and TG, and, as silencing caused no reduction in the 46 kDa band and scr-siRNA affected neither of the two bands, show that in U937 cells the size of JNK2 proteins is 54 kDa, and that these were upregulated and activated after ER stress. The 46 kDa band, on the other hand, is made up of constitutively expressed JNK proteins, perhaps explaining why they were more difficult to silence. The fact that both TN and TG were shown to increase JNK2 levels and the possibility to use efficiently silenced cells prompted us to investigate its role in U937 cell survival.
JNK2 regulates BiP and CHOP
Disturbance of ER function leads to an accumulation of unfolded proteins and activation of the UPR. Numerous studies have shown that survival and death decisions during the UPR are, respectively, mediated via antiapoptotic BiP and proapoptotic CHOP. We therefore examined the expression of these proteins by western blot after JNK inhibition by anthra[1,9-cd]pyrazol-6(2H)-one (SP600125), used at the concentration of 10 μM, which is known to inhibit specifically these stress-activated protein kinases (SAPK).29 TN caused an increase in both BiP and CHOP, which became evident after 6 h and marked after 9 and 12 h. JNK inhibition caused a change in their expression over time – the increase of CHOP became more pronounced after 6, 9 and 12 h, while BiP levels declined, even in the absence of TN, suggesting that JNK basal activity may be involved in the regulation of the expression of this protein (Figure 3a). As SP600125 inhibits both JNK1 and JNK2, to further evaluate the relationship between JNK2 and the UPR, BiP and CHOP were analyzed by western blot after JNK2 silencing. Transfection time was 24 h, and TN or TG were added during the subsequent 6 h. Irrespective of which of the two stressors was added, JNK2 silencing imposed a decrease in BiP of about 40–50% and a four- to sixfold increase in CHOP, in comparison with scr-siRNA (Figure 3b). These results indicate that JNK2 is linked to UPR regulation favoring a prosurvival profile.

Effect of JNK inhibition or silencing on BiP and CHOP levels. (a) U937 cells were pretreated for 30 min with SP600125 (10 μM) or not before stressing with TN (1 μM) or not for 6, 9 or 12 h. In the samples reported as 0 h, TN was absent and SP600125 was either absent or present for 12 h. Western blot was performed to analyze BiP and CHOP levels. Uniform loading of proteins was confirmed by β-actin probing in the same samples. Representative blots out of three experiments are shown. (b) Involvement of JNK1 and JNK2 in BiP and CHOP expression after TN or TG stress was investigated by JNK1- and JNK2-siRNA, respectively, whose specificities were checked by transfecting U937 cells with scr-siRNA. Cells were transfected for 24 h, and TN (1 μM) or TG (200 nM) was added for a further 6 h. Uniform protein loading was confirmed by probing β-actin in the same samples. Densitometric quantification is shown at the bottom of each line as the ratio of BiP or CHOP values in the lysates of JNK1- or JNK2-siRNA-transfected cells relative to BiP or CHOP values in the scr-siRNA sample. This type of experiment was performed twice with comparable results
JNK2 regulates the cellular acidic/lysosomal cellular compartment
Transmission electron microscopy (TEM) examination of the ultrastructural features of untreated U937 cells revealed well-preserved and morphologically normal intracellular organelles, specifically the mitochondria, rough ER (rER) and Golgi apparatus, along with sparse electron-dense structures, identifiable as lysosomes or autolysosomes (Figure 4, inset 1). After 20–24 h, and in fact after 14 h of TN treatment, the rER appeared dilated, indicating ER stress, and the mitochondria displayed dilated cristae. In addition, a marked increase in autolysosomes, associated with vacuolar compartments surrounded by single or double membranes, morphologically consistent with endosomes and autophagosomes, was observed in the cytoplasm (Figure 4, inset 2, 5 and 8). These structures were less evident in cells treated with 3-methyladenine (3-MA) (Figure 4, inset 3), an inhibitor of the earliest stages of autophagosome formation. However, they were present after pharmacological inhibition of JNK by SP600125 (Figure 4, inset 4), and the number of autolysosomal structures was shown to increase in cells pretreated with SP600125 and then exposed to TN (Figure 4, inset 4, 6, 7 and histogram). In this case, the ultrastructural aspects observed after 14 h changed radically at later time intervals, and after 20–24 h, the apoptotic features completely dominated the picture (Figure 4, inset 9 and 10).

JNK inhibition induces accumulation of electron-dense structures. Conventional ultrastructural analysis of U937 cells either treated or untreated with TN (1 μM) in the presence or absence of SP600125 (insets 1–10). Cells treated with TN for 20 h (inset 2) showed, in comparison to control (inset 1), dilated ER and cytoplasmic vacuolizations corresponding to typical acidic compartments, ranging from endosomes to lysosomes (inset 2, arrow) and including autophagic vacuoles (inset 5) and autolysosomes (inset 8). Cells exposed for 20 h to both TN and the autophagy inhibitor 3-MA (5 mM) showed only dilated ER (inset 3). Pharmacological inhibition of JNK with SP600125 (10 μM) in TN-treated cells increased the accumulation of electron-dense autolysosomal structures (inset 4) and autophagic vacuoles at 14 h (inset 4, 6 and 7). Apoptotic bodies were evident at 20 h treatment (inset 9 and 10). Bars: 1 mm for insets 1–4 and 0.1 mm for insets 5–8. The histogram shows the quantitative analysis of the experiment in insets 1–10, performed by counting electron-dense autolysosomes in 30 different cells for each treatment. Values are means±S.D.
To detect the role of JNK in the acidic compartment, U937 cells were stained with acridine orange (AO), a weak base that accumulates in acidic spaces and fluoresces bright red. TN induced an increase in the red fluorescence in comparison with untreated cells (Figure 5a), and SP600125 (10 μM) and JNK2-specific inhibitor IX (50 nM)30pretreatments both caused a further increase in the bright red fluorescence (Figure 5a). As expected, the autophagy inhibitor 3-MA prevented the development of the red fluorescence, which, instead, was increased by chloroquine, an inhibitor of the degradation phase of autophagy, as this inhibits lysosomal acidification and prevents autophagosome–lysosome fusion (not shown). AO red fluorescence was also analyzed by cytofluorimetry after JNK2 silencing, and a greater fluorescence was detected in these cells, in comparison with scr-siRNA-transfected cells, upon TN treatment (Figure 5b).

JNK2 inhibition or silencing induces an increase in the acidic compartment. (a) AO and (c) LysoTracker Red staining of cells treated or untreated with TN (1 μM), either in the presence or absence of SP600125 (10 μM) or inhibitor IX (50 nM), for 14 h. Arrows indicate areas of accumulation of acidic compartments in cells analyzed under immunofluorescence microscopy. (b) U937 cells, transfected with JNK2-siRNA or scr-siRNA for 24 h and exposed or not to TN (1 μM) for further 14 h, were stained with AO and analyzed by cytofluorimetry. The mean fluorescence intensities were measured from the histograms representing FL2 (log) in ≥10 000 events. The results are expressed according to the formula (MFI in JNK2-siRNA-treated cells)/(MFI in scr-siRNA-treated cells) × 100. The values of one of two independent experiments, which yielded comparable results, are shown. (d) Cells treated and stained with LysoTracker Red, as described in panel c, were analyzed by flow cytometry, and the mean fluorescence intensities were measured from the histograms representing FL2 (log) in ≥10 000 events. Data are the means±S.D. of three independent experiments. Statistical analysis by Student’s t-test is shown
U937 cells were also stained with LysoTracker Red DND-99, a fluorescent acidotropic probe, which is more selective for acidic organelles. As expected from the ultrastructural aspects and AO staining, LysoTracker Red fluorescence was more pronounced in cells treated with TN in comparison with untreated cells, and a larger accumulation of the dye was evident after JNK inhibition with SP600125 or JNK2-specific inhibitor IX (Figure 5c). LysoTracker Red fluorescence was also evaluated by cytofluorimetry, and TN treatment increased the red fluorescence of the acidic organelles by about 30% in comparison with untreated cells. When TN was added after JNK inhibition by either SP600125 or by inhibitor IX, this rose to 50–60% (Figure 5d). Similar results were obtained when the ER stress was induced by TG (not shown).
These experiments indicate that ER stressors generate an increase in the acidic compartment that is even more sustained when JNK2 is inhibited or silenced, suggesting a regulatory role of this SAPK. However, the experiments performed were not able to clarify the type of JNK2 involvement. Indeed, without further experimentation it would be impossible to state whether the increase in the acidic compartment observed depended on its active formation or, instead, its accumulation caused by a functional or structural impairment.
JNK2 regulates the final steps of TN-induced autophagy
ER stress may lead to upregulation of the autophagic machinery as a survival mechanism.7, 8, 9, 10 Therefore, to evaluate a possible role of JNK2 as regulator of this complex and multistep process, we first analyzed microtubule-associated protein light chain 3 (LC3) in TN-treated cells by western blot. This revealed a time-dependent increase in phosphatidylethanolamine-conjugated microtubule-associated protein 1 light chain 3 (LC3-II) (Figure 6a), suggesting that TN treatment increases the number of autophagosomes. As LC3-II increase could be caused either by enhanced autophagic sequestration or by reduced degradation of autophagic material, we also analyzed p62, as this protein, being degraded in the final autophagic steps, is used as an autophagic flux readout.31 By western blot it was observed that cells in which JNK2 was blocked by inhibitor IX did not show the p62 decrease caused by TN treatment alone (Figure 6b). Furthermore, JNK2 silencing, but not scr-siRNA, caused an increase of p62 after TN cell treatment (Figure 6c). These results indicate that JNK2 has a role to play in the regulation of the final degradative steps, thus avoiding autolysosomes and lysosomes accumulation.

JNK2 regulates the final steps of autophagy. (a) TN (1 μM) induces an increased autophagosomes formation, as detected by western blot analysis of LC3-I/II; (b) TN induces an autophagic flux, blocked by JNK2 inhibitor IX (50 nM) and (c) by JNK2 silencing, but not by scr-siRNA transfection. In the last experiment, the cells were transfected for 24 h and exposed or not to TN (1 μM) for 14 h. The experiment reported was repeated three times and the experiments in (b) and in (c) were repeated twice with comparable results. The level of β-actin is shown at the bottom of each blot as a loading control
JNK2 counters apoptosis
TEM showed that JNK inhibition in TN-treated cells causes an increase in the acidic compartment after 14 h, but striking apoptotic features prevailed if this treatment was prolonged to 24 h (Figures 4 (inset 9 and 10) and 7a). Apoptosis was confirmed in both TN- and TG-stressed cells in separate experiments in which JNK2 was inhibited by SP600125 or by inhibitor IX. In detail, upon TN-delivered stress, JNK inhibition by SP600125 caused 53±5% of Annexin V-positive cells and 48±3% of sub-G1 events (Figures 7b and c). Similar findings were obtained with TG as stressor: Annexin V-positive cells were 44±1% and sub-G1 events were 41±5%. When JNK2 was inhibited by inhibitor IX in TN-stressed cells, the fraction of Annexin V-positive cells was 60±7% and sub-G1 events were 47±7%. Similar results were observed when TG was used as the stressor (Figures 7b and c). Moreover, 3-MA, an autophagy inhibitor, induced an increase in apoptotic death upon both TN and TG treatments (Figures 7b and c). Under the same conditions, neither the inhibitors nor DMSO affected cell viability (not shown). Upon JNK pharmacological inhibition, caspase 3 was found to be fragmented and therefore activated, after 18 h of TN treatment (Figure 7d). Apoptotic cell death was also consistently enhanced following TN treatment in JNK2-silenced cells, as detected by Annexiv V cell binding (Figure 7e). Hence, the results obtained with both pharmacological inhibitors, and further confirmed by JNK2 silencing, allow us to conclude that JNK2 has an important role in the survival pathway induced by ER stress.

JNK2 inhibition or lack of expression induces apoptosis in TN-stressed cells. (a) Apoptotic bodies observed by TEM at 24 h treatment with TN and SP600125 (10 μM). (b and c) Detection of cell death by evaluation of sub-G1 events (b) and Annexin V binding (c). U937 cells were pretreated for 30 min with SP600125 (10 μM), inhibitor IX (50 nM), 3-MA (5 mM) or not, and then with TN (1 μM) or TG (200 nM) or not for 24 h. A portion of cells were fixed and stained with PI to evaluate sub-G1 events in the cell cycle under cytofluorimetry, and another portion of cells was used to detect the binding of FITC-conjugated Annexin V by cytofluorimetry. Both types of investigation were performed on ≥10 000 events. The values reported are means±S.D. (n=5). Assessment of cell death showed statistically significant differences (*P<0.05) between the data obtained in the cultures treated with TN or TG, together with their respective inhibitors, in comparison with the cultures treated with TN or TG alone. (d) Cell death by apoptosis was confirmed by western blot detection of caspase 3 fragmentation in cells pretreated for 30 min with SP600125 (10 μM) and then with TN (1 μM) for the times indicated. In the 0-h samples, TN was absent, whereas SP600125 was either absent or present for 24 h. As positive control of caspase fragmentation, a lysate of U937 cells treated with TNFα was included in the same blot. (e) U937 cells transfected with JNK2-siRNA or scr-siRNA for 24 h and exposed or not to TN for 24 h were analyzed under cytofluorimetry to detect the binding of FITC-conjugated Annexin V. Here are shown the values of one of two independent experiments, which yielded comparable results
Discussion
Although extensive studies have been conducted, stress-induced JNK activation is still a matter of debate. Indeed, it appears to be implicated in the regulation of contrasting cellular processes, such as proliferation and survival on the one hand and apoptosis on the other.7, 8, 20, 21, 22, 23, 24 Nevertheless, the discovery of different modes of JNK activation may provide a partial explanation to this appparent paradox. In fact, it may be that while its early and transitory activation signals for cell survival, its sustained activation triggers apoptosis.27 Even though the functional significance of the different JNK isoforms in human cells is far from clear, JNK1 and JNK2 have been reported as functionally distinct. This indicates that they may have selective roles in mediating stress-induced apoptosis and regulating gene expression.25, 26
In the present investigation, we show that, in U937 cells, moderate ER stress generated by non-cytotoxic concentrations of TN or TG leads to early transitory activation of JNK, migrating as a 54 kDa band made up of JNK2 isotypes, as this could be completely silenced by JNK2-specific siRNA. Although JNK1 and JNK2 were expected to display 46 and 54 kDa isotypes, it is apparent from our findings in U937 cells that JNK2 is synthesized as 54 kDa protein(s), as previously reported in HeLa cells, mouse 3T3 fibroblasts and other cell types.32, 33 Both pharmacological inhibition and expression silencing, by specific siRNA, revealed a prosurvival role of this SAPK in ER-stressed cells.
ER stress-activated gene transcription is mediated by three different but interconnected pathways: PERK-ATF-4, ATF6 and IRE-JNK/XBP1. The transducers of these pathways (PERK, ATF6 and IRE1) sense the presence of unfolded proteins in the ER lumen and convey this information to the nucleus, and in the case of IRE1, lead to JNK activation and autophagy induction.7, 8 Although we did not set out to explore the link between IRE1 and JNK, we were able to pinpoint the role of JNK2 in ER stress regulation. Like previous studies, reporting that JNK2 causes BiP upregulation and protection from death,34, 35 we observed a JNK2-dependent increase in the expression of BiP, and, furthermore, we detected that the same SAPK counters CHOP expression. Indeed, both pharmacological inhibition and siRNA silencing of JNK2 altered the time kinetics of both BiP and CHOP expression, delaying the increase in the former and promoting early, more sustained upregulation in the latter. This seems to suggest that JNK2 has a role in a survival pathway potentially featuring BiP and CHOP, as the former is antiapoptotic and prevents ER stress-induced cell death,3, 4 while the latter is a transcription factor implicated in apoptosis and in cell death.5, 6
ER is not only involved in protein synthesis and quality control but also represents a major source of the autophagy isolation membranes.10, 11 As ER stress can affect autophagic flux, we can hypothesize that this may be impaired by JNK inhibition, particularly since JNK has already been shown to increase autophagy.7, 8 Although it is true that others ascribe JNK a role in autophagy suppression, as demonstrated in neurons,36 our experiments indicate that JNK2 has a specific role as a regulator of the cellular acidic compartment, preventing its accumulation in ER-stressed cells undergoing autophagic flux. These conclusions were reached following western blot analysis of p62 in cells undergoing autophagic flux following TN-induced ER stress. This molecule increased after JNK2 silencing and failed to decrease after JNK2 pharmacological inhibition, thereby indicating that the increase in acidic vacuoles and lysosomes observed was due to their accumulation rather than a rise in their formation. Nonetheless, the fate of autolysosomes is not entirely clear. It is currently thought that autophagy ends up with degradation of the autophagosome cargo in autolysosomes, and that this process involves nutrient regeneration.37, 38 Indeed, a lysosome reformation mechanism has been detected following autolysosome induction by starvation, and found to depend on mTOR reactivation, autolysosomal protein degradation and functioning microtubules. Inhibition of each of these steps blocks autophagic lysosome reformation and instead induces the formation and persistence of giant lysosomes.39 Other studies have shown that moderate lysosomal rupture induces apoptosis, whereas severe lysosomal leakage results in necrosis without caspase activation.17, 18, 40, 41 Our contribution to this body of work appears to suggest that subcytotoxic ER stress transiently activates JNK2, bringing about degradation through the autophagic flux of damaged proteins and organelles. This leads us to conclude that JNK2, along with BiP and CHOP, can regulate other UPR proteins responsible for maintaining autolysosome and lysosome function and, by this mechanism, promote cell survival. As ER stress, autophagy and lysosomes are known to be involved in senescence, neurodegenerative diseases and cancer cell responses to chemotherapy, JNK2 could therefore be a potentially enticing therapeutic target.
Materials and Methods
Materials
AO, anti-β-actin antibody, bovine serum albumin (BSA), chloroquine, fetal calf serum (FCS), Hanks’ balanced salt solution (HBSS), L-glutamine, 3-MA, penicillin–streptomycin, phosphate-buffered saline (PBS), PI, RNAse, RPMI 1640 and TN were from Sigma-Aldrich (St Louis, MO, USA). Anisomycin, SP600125 and TG were from Calbiochem (Inalco, Milan, Italy) and JNK2/3 inhibitor IX was from Merck (INALCO, Milan, Italy). Antibodies anti-pSAPK/JNK (Ser473), -JNK, -BiP, -CHOP and -LC3-I/II were from Cell Signaling Technology (Beverly, MA, USA). Anti-caspase 3 antibody siRNA for human JNK1, JNK2 and non-silencing scr-siRNA were from Santa Cruz Biotechnology (Tebu-Bio, Magenta, Italy). Mouse anti-p62 was from BD Biosciences (San Jose, CA, USA) and Lipofectamine RNAiMAX, OPTI-MEM medium and LysoTracker Red DND-99 were from Invitrogen (San Giuliano Milanese, Italy). Horseradish peroxidase-conjugated anti-rabbit- and anti-mouse-immunoglobulin antibodies, enhanced chemiluminescence (ECL) reagents and Hyperfilm-ECL film were from Amersham (Arlington Heights, IL, USA), and RC DC protein assay, sodium dodecyl sulfate (SDS)-sample buffer, protein standard, SDS-polyacrylamide gel electrophoresis (SDS-PAGE) reagents and polyvinylidene difluoride (PVDF) membranes were from Bio-Rad Laboratories (Segrate, Italy). Fluorescein isothiocyanate (FITC)-labeled Annexin V was from ImmunoTools GmbH (Altenoyther, Germany), and other reagents were of the highest purity and purchased from Bio-Rad (Segrate, Milan, Italy), Invitrogen or Sigma.
Cell viability and growth
The human monoblastic U937 cells were grown in complete medium (RPMI-1640 medium supplemented with 5% heat-inactivated FCS, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin) at 37 °C, in fully humidified 95% room air/5% CO2. Cells were resuspended three times a week in fresh complete medium to 3 × 105/ml. Cell growth was evaluated by hemocytometry counts of cells excluding Trypan blue (0.04% Trypan blue in PBS (w/v)), and viability was assessed by calculating alive (Trypan blue excluding) cells as percentage of all cells counted. Cells used in every experiment were ≥94% viable and taken from cultures in exponential growth. They were washed once and resuspended in complete medium, 1 × 106/ml, and transferred to 24-well microplates. They were then treated with inhibitors or vehicles, incubated for 30 min, and subsequently exposed to test agents or, again, to vehicles. At the end of each experiment, the cells were gently mixed and aliquots were taken for cell counting and cell cycle analysis. The vehicles, even when used in combination, were ≤0.3% (v/v) and did not modify any investigated parameter in comparison with control cultures.
Flow cytometry analysis of cell death, phosphatidylserine exposure and cell membrane permeability
Nuclear DNA fragmentation was quantified by flow cytometry of hypodiploid (sub-G1) DNA after cell fixation and PI staining.28, 42 Briefly, cells were washed with PBS, pelletted and fixed in ice-cold ethanol/water (70/30, v/v) for 1 h, pelletted again and washed twice with PBS, and finally resuspended in PBS containing RNAse (20 μg/ml) and PI (100 μg/ml). Events in the different cell cycle phases were gated manually using an EPICS XL cytofluorimeter (Beckman Coulter, Hialeah, FL, USA). At least 10 000 events per sample were acquired. Collected data were analyzed using the Multicycle software for DNA content and cell cycle analysis (Phoenix Flow System, San Diego, CA, USA). The sub-G1 events representative of the apoptotic cells, and the events in the other cell cycle phases, are given as a percentage of the total cell population. Apoptotic cells were detected by staining with FITC-conjugated Annexin V and PI. Briefly, 2.0 × 105 U937 cells were washed and resuspended in 200 μl of Annexin V-binding buffer (HEPES, 10 mM, pH 7.4, CaCl2, 2.5 mM, MgCl2, 1 mM, KCl, 5 mM, NaCl, 140 mM) containing PI (200 μg/ml) and FITC-Annexin V, according to the manufacturer’s instructions. Gently mixed cells were incubated for 15 min at room temperature in the dark. A measure of 300 μl of Annexin V-binding buffer were added to all samples, which were then kept on ice and analyzed within 1 h by flow cytometry (EPICS-XL), measuring the fluorescence emission at 530 nm (FL1) and >575 nm (FL3). Cells labeled by Annexin V were considered apoptotic and Annexin V- and PI-negative cells were considered viable.
Western blot analysis
Whole cell lysates were prepared as described previously.43 Briefly, the cells were kept for 30 min on ice in lysis buffer (NaCl 150 mM, CaCl2 1 mM, MgCl2 1 mM, NaN3 0.1%, NaF 10 mM, Triton X-100 1% (v/v), orthovanadate 1 mM, aprotinin 2 μg/ml, leupeptin 2 μg/ml, iodoacetamide 10 mM, PMSF 2 mM and pepstatin 20 μM). The appropriate volumes of 4 × SDS-sample buffer and 2-mercaptoethanol 5% (v/v) were then added. Cell lysates were briefly sonicated, warmed at 95 °C for 5 min and cleared by 14 000 × g centrifugation in a microfuge for 15 min at 4 °C. Supernatants were collected and proteins were quantified by RC DC protein assay. Equal amounts of proteins were separated from the different samples by SDS-PAGE, and blotted onto PVDF membranes. Transfer efficiency was checked with Ponceau staining. The blots were blocked in Tris-buffered saline containing BSA 2% (w/v), probed with specific primary antibodies, washed with PBS-Tween-20 and then incubated with a peroxidase-conjugated secondary antibody. Finally, each membrane was probed to detect β-actin. The final dilutions and incubation times suggested by the manufacturer were used for each antibody. Immunodetection was performed using the ECL reagents and Hyperfilm-ECL film. Densitometry quantitation of the bands was performed using the ImageJ software (National Institutes of Health, Bethesda, MD, USA) on a Mac OS 9.0 (Apple Computer International, Cupertino, CA, USA).
siRNA
RNA knockdown was performed with pools of siRNA duplexes. Briefly, cells were washed and resuspended in OPTI-MEM medium, transfected with siRNA corresponding to human JNK1 or JNK2 and with scr-siRNA, using Lipofectamine RNAiMAX according to the manufacturer’s guidelines. A parallel cell sample was transfected with a fluorescent siRNA. After 7 h of incubation, RPMI 1640 containing 20% normal calf serum was added without removing the transfection medium. The cells were cultured for further 24 h. After centrifugation, the medium was replaced with fresh RPMI 1640, containing 10% FCS, and the cells cultured again in the presence of either TN or TG or not.
Electron microscopy
Cells were collected and fixed in 4% paraformaldehyde and 2% glutaraldehyde in PBS for 2 h at room temperature, post-fixed with 1% OsO4 for 2 h and stained for 1 h in 1% aqueous uranyl acetate. The samples were then dehydrated with graded acetone and embedded in Epon-812 (Electron Microscopy Science, Societa' Italiana Chimici, Rome, Italy). Ultrathin sections were cut using a Reichert ultramicrotome, counterstained with uranyl-acetate and lead citrate, and examined under a Philips CM10 transmission electron microscope (FEI, Eindhoven, The Netherlands).
Acidic compartment evaluation by AO
Autophagy is characterized by the formation of acidic vesicular organelles (autophagosomes and autolysosomes). AO, a fluorescent weak base, causes these acidic compartments to fluoresce bright red, and the cytoplasm and nucleolus to fluoresce bright green and dim green, respectively. AO staining (100 μg/ml) was performed in the dark for 30 min at room temperature after cell pellets were collected and resuspended in 1 ml of HBSS, supplemented with 5% FCS. Staining was performed in the presence of the different drugs or their vehicles. Cells were then washed twice with HBSS, resuspended in the same medium, spotted on glass coverslips and analyzed using an Apotome Axio Observer Z1 inverted microscope (Zeiss, Oberkochen, Germany) equipped with an AxioCam MRM Rev.3, at × 40 and × 63 magnification. Fluorescence signal was analyzed using the AxioVision4.6.3 software (Carl Zeiss, Germany), and image analysis was performed by means of Adobe Photoshop.
As for the cytofluorimetric analysis, JNK2-silenced and scr-siRNA-treated cells were either exposed or not to TN for 14 h, stained as described above, and analyzed in a cytofluorimeter (EPICS XL, Beckman Coulter). Cells showed similar side- and forward-scatter characteristics. The gate used for each sample was the same as in control cells, and the red fluorescence intensity of the gated cells was analyzed on a log scale (Fl2) and recorded as mean fluorescence intensity (MFI). A minimum of 10 000 events were examined for each sample. The results are expressed according to the formula (MFI in JNK2-siRNA-treated cells)/(MFI in scr-siRNA-treated cells) × 100.
Acidic compartment evaluation by LysoTracker Red
Cells were incubated for 15 min in HBSS containing LysoTracker Red DND-99 (100 nM), a fluorescent acidotropic probe with high selectivity for acidic organelles, showing good retention after aldehyde fixation. HBSS-washed cells were either analyzed in a cytofluorimeter EPICS XL or fixed with paraformaldehyde (2%) in PBS for 30 min at room temperature and then analyzed using an Apotome Axio Observer Z1 inverted microscope (Zeiss), equipped with an AxioCam MRM Rev.3, at × 40 and × 63 magnification. Fluorescence signal was analyzed using the AxioVision4.6.3 software and image analysis was performed by means of Adobe Photoshop.
Under cytofluorimetry, control and treated cells showed similar side- and forward-scatter characteristics; the gate used for each sample was the same as in control cells. The red fluorescence intensity of the gated cells was analyzed on a log scale (Fl2) and recorded as MFI. A minimum of 10 000 events were examined for each sample, and the results are expressed according to the formula (MFI in treated cells)/(MFI in control cells) × 100.
Statistical analysis
Results are expressed as the mean±standard deviation (S.D.) of repeated experiments, as indicated in the figure legends. Statistical differences were evaluated using paired two-tailed Student’s t-test. Differences were considered statistically significant for values of P≤0.05.
Abbreviations
- AO:
-
acridine orange
- BiP:
-
immunoglobulin heavy-chain binding protein
- BSA:
-
bovine serum albumin
- CHOP:
-
CCAAT/enhancer-binding protein-homologous protein
- ECL:
-
enhanced chemiluminescence
- ER:
-
endoplasmic reticulum
- FCS:
-
fetal calf serum
- FITC:
-
fluorescein isothiocyanate
- GRP78:
-
78 kDa glucose-regulated protein
- HBSS:
-
Hanks’ balanced salt solution
- IRE1:
-
inositol-requiring kinase 1
- JNK:
-
c-Jun N-terminal kinase
- LC3-I:
-
microtubule-associated protein 1 light chain 3
- LC3-II:
-
phosphatidylethanolamine-conjugated microtubule-associated protein 1 light chain 3
- 3-MA:
-
3-methyladenine
- MAPK:
-
mitogen-activated protein kinase
- MFI:
-
mean fluorescence intensity
- MKK:
-
mitogen-activated protein kinase kinase
- PBS:
-
phosphate-buffered saline
- PI:
-
propidium iodide
- PVDF:
-
polyvinylidene difluoride
- SAPK:
-
stress-activated protein kinases
- SDS-PAGE:
-
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- siRNA:
-
small interfering RNA
- SP600125:
-
anthra[1,9-cd]pyrazol-6(2H)-one
- TEM:
-
transmission electron microscopy
- TG:
-
thapsigargin
- TN:
-
tunicamycin
- UPR:
-
unfolded protein response
References
Kaufman RJ . Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 1999; 13: 1211–1233.
Harding HP, Calfan M, Urano F, Novoa I, Ron D . Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol 2002; 18: 575–599.
Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J . The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature (London) 1988; 332: 462–464.
Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res 2008; 68: 498–505.
Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ et al. Signals from the stressed endoplasmic reticulum induce C/EBP homologous protein (CHOP/GADD153). Mol Cell Biol 1996; 16: 4273–4280.
Marciniak SJ, Yun CY, Oyadamari S, Novoa I, Zhang Y, Jungreis R et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004; 18: 3066–3077.
Ogata M, Hino SI, Saito A, Morikawa K, Kondo S, Kanemoto S et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26: 9220–9231.
Ding W-X, Ni H-M, Gao W, Yoshimori T, Stolz DB, Ron D et al. Linking of autophagy to ubiquitin–proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171: 513–524.
Yorimitsu T, Nair U, Yang Z, Klionsky DJ . Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281: 30299–30304.
Bernales S, McDonald KL, Walter P . Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 2006; 4: 2311–2324.
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182: 685–701.
Alva AS, Gultekin SH, Baehrecke EH . Autophagy in human tumors: cell survival or death? Cell Death Differ 2004; 11: 1046–1048.
Gozuacik D, Kimchi A . Autophagy and cell death. Curr Top Dev Biol 2007; 78: 217–245.
Eskelinen EL, Tanaka Y, Saftig P . At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol 2003; 13: 137–145.
Luzio JP, Pryor PR, Bright NA . Lysosomes: fusion and function. Nat Rev Mol Cell Biol 2007; 8: 622–632.
Joyce JA, Hanahan D . Multiple roles for cysteine cathepsins in cancer. Cell cycle 2004; 3: 1516–1519.
Guicciardi ME, Leist M, Gores GJ . Lysosomes in cell death. Oncogene 2004; 23: 2881–2890.
Kroemer G, Jäättelä M . Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5: 886–897.
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000; 287: 664–666.
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B . JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30: 678–688.
Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009; 28: 886–898.
Oh SH, Lim SC . Endoplasmic reticulum stress-mediated autophagy/apoptosis induced by capsaicin (8-methyl-N-vanillyl-6-nonenamide) and dihydrocapsaicin is regulated by the extent of c-Jun NH2-terminal kinase/extracellular signal-regulated kinase activation in WI38 lung epithelial fibroblast cells. J Pharmacol Exp Ther 2009; 329: 112–122.
Shimizu S, Konishi A, Nishida Y, Mizuta T, Yamamoto A, Tsujimoto Y . Involvement of JNK in the regulation of autophagic cell death. Oncogene 2010; 29: 2070–2082.
Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K . Reactive oxygen species mediate crosstalk between NF-κB and JNK. Cell Death Differ 2006; 13: 730–737.
Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Dérijard B et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 1996; 15: 2760–2770.
Bogoyevitch MA . The isoform-specific functions of the c-Jun N-terminal kinases (JNKs): differences revealed by gene targeting. BioEssays 2006; 28: 923–934.
Ventura JJ, Hübner A, Zhang C, Flavell RA, Shokat KM, Davis RJ . Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell 2006; 21: 701–710.
Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C . A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 1991; 139: 271–279.
Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 2001; 98: 13681–13686.
Snider JL, Allison C, Bellaire BH, Ferrero RL, Cardelli JA . The β1 integrin activates JNK independent of CagA, and JNK activation is required for Helicobacter pylori CagA+-induced motility of gastric cancer cells. J Biol Chem 2008; 283: 13952–13963.
Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askow DS et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4: 151–175.
Gancz D, Donin N, Fishelson Z . Involvement of the c-jun N-terminal kinases JNK1 and JNK2 in complement-mediated cell death. Mol Immunol 2009; 47: 310–317.
Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF . Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-jun-dependent cell proliferation. Mol Cell 2004; 15: 713–725.
Potapova O, Anisimov SV, Gorospe M, Dougherty RH, Gaarde WA, Boheler KR et al. Targets of c-Jun NH2-terminal kinase 2-mediated tumor growth regulation revealed by serial analysis of gene expression. Cancer Res 2002; 62: 3257–3263.
Potapova O, Gorospe M, Dougherty RH, Dean NM, Gaarde WA, Holbrook NJ . Inhibition of c-Jun N-terminal kinase 2 expression suppresses growth and induces apoptosis of human tumor cells in a p53-dependent manner. Mol Cell Biol 2000; 20: 1713–1722.
Xu P, Das M, Reilly J, Davis RJ . JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 2011; 25: 310–322.
Jäger S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004; 117: 4837–4848.
Jahreiss L, Menzies FM, Rubinsztein DC . The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008; 9: 574–587.
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465: 942–946.
Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 2010; 17: 1167–1178.
Boya P, Kroemer G . Lysosomal membrane permeabilization in cell death. Oncogene 2008; 27: 6434–6451.
Matusali G, Arena G, De Leo A, Di Renzo L, Mattia E . Inhibition of p38 MAP kinase pathway induces apoptosis and prevents Epstein–Barr virus reactivation in Raji cells exposed to lytic cycle inducing compounds. Mol Cancer 2009; 8: 18.
Marfè G, Morgante E, Di Stefano C, Di Renzo L, De Martino L, Iovane G et al. Sorbitol-induced apoptosis of human leukemia is mediated by caspase activation and cytochrome c release. Arch toxicol 2008; 6: 371–377.
Acknowledgements
We thank Prof. Renato Mariani-Costantini for his helpful comments and suggestions. This work was supported by grants to LDR from Sapienza Ateneo 2010 and 2011 (8.1.1.1.32.5 and 8.1.1.1.34.1).
Author contributions
MR performed most of the experiments, participated in the study design and analyzed the results. LVL performed, analyzed and wrote the morphological studies, and advised on the research. SV performed experiments and analyzed results. FMP participated in the analysis of the results, while LDR designed the experiments, analyzed the data, coordinated the study group and wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Raciti, M., Lotti, L., Valia, S. et al. JNK2 is activated during ER stress and promotes cell survival. Cell Death Dis 3, e429 (2012). https://doi.org/10.1038/cddis.2012.167
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.167
Keywords
This article is cited by
-
JNK1 and JNK3: divergent functions in hippocampal metabolic-cognitive function
Molecular Medicine (2022)
-
Intrinsic disorder in proteins associated with oxidative stress-induced JNK signaling
Cellular and Molecular Life Sciences (2022)
-
Depletion of eukaryotic initiation factor 5B (eIF5B) reprograms the cellular transcriptome and leads to activation of endoplasmic reticulum (ER) stress and c-Jun N-terminal kinase (JNK)
Cell Stress and Chaperones (2021)
-
Defining the underlying defect in insulin action in type 2 diabetes
Diabetologia (2021)
-
The Jun N-terminal kinases signaling pathway plays a “seesaw” role in ovarian carcinoma: a molecular aspect
Journal of Ovarian Research (2019)
