
Abstract
Oxidative stress leads to T-cell hyporesponsiveness or death. The actin-binding protein cofilin is oxidized during oxidative stress, which provokes a stiff actin cytoskeleton and T-cell hyporesponsiveness. Here, we show that long-term oxidative stress leads to translocation of cofilin into the mitochondria and necrotic-like programmed cell death (PCD) in human T cells. Notably, cofilin mutants that functionally mimic oxidation by a single mutation at oxidation-sensitive cysteins (Cys-39 or Cys-80) predominately localize within the mitochondria. The expression of these mutants alone ultimately leads to necrotic-like PCD in T cells. Accordingly, cofilin knockdown partially protects T cells from the fatal effects of long-term oxidative stress. Thus, we introduce the oxidation and mitochondrial localization of cofilin as the checkpoint for necrotic-like PCD upon oxidative stress as it occurs, for example, in tumor environments.
Similar content being viewed by others


Main
The balance between T-cell activation and T-cell inactivation is important for physiological immune responses. The microenvironment has a decisive part in controlling the T-cell-mediated immune response. Oxidative stress, for example, induces T-cell hyporesponsiveness, which is harmful in cancer settings,1 but auxiliary in controlling autoimmune diseases, for example, arthritis.2 In the latter case, production of reactive oxygen species (ROS), especially through neutrophils and macrophages, is thought to control self-reactive T cells. Thus, the release of ROS is not only per se harmful, but also an important control mechanism for T-cell-mediated immune responses.
T cells produce low amounts of hydrogen peroxide (H2O2) upon TCR triggering, which even facilitates T-cell activation.3 However, T cells can only cope with a certain amount of ROS. High concentrations of pro-oxidants, for example, H2O2, ultimately lead to T-cell hyporesponsiveness4 or death.5 The molecular mechanisms that control the T-cell fate upon ROS exposure are largely unknown. Kaminski et al.6 showed that the mitochondria has a role in ROS-mediated upregulation of FasL. We have recently shown that cofilin is a direct target of ROS in primary human T cells.4 Here we show that in oxidative stress conditions, oxidation of cofilin leads to its mitochondrial translocation and induces necrotic-like programmed cell death (PCD).
Cofilin is a key regulator in dynamic reorganization of the actin cytoskeleton during T-cell activation. The binding of cofilin to actin leads to depolymerization or severing of existing actin filaments. The latter allows elongation of the newly generated actin filaments and thus a net actin polymerization. Activation of cofilin is required for formation of the immune synapse and T-cell activation (reviewed in Samstag et al.7). If cofilin is oxidized, it will no longer be able to depolymerize actin filaments, although it still associates with F-actin. Mechanistically, cofilin oxidation induces a conformational change and the loss of its phosphorylation.4 The inability of oxidized cofilin to control actin dynamics eventually leads to a stiff actin cytoskeleton and concomitantly to T-cell hyporesponsiveness. Therefore, T-cell hyporesponsiveness induced by oxidative stress can at least in part be explained by cofilin oxidation.
Although it is well accepted that oxidative stress can induce T-cell death, there are ambiguous data with regard to the type of death the cells are committed to.5, 8 Here, we show that long-term oxidative stress induces the mitochondrial localization of cofilin, which is sufficient to induce necrotic-like PCD. Accordingly, knockdown of cofilin protects T cells in part against the detrimental effects of H2O2. Thus, oxidation and mitochondrial transport of cofilin represent checkpoints that determine T-cell fate in oxidative stress conditions.
Results
Long-term oxidative stress leads to caspase-independent death of primary human T cells
To analyze cell death induction through exposure of ROS in primary human T cells, we first compared T cells incubated with 100 μM H2O2 for 2 h (short term) with T cells incubated for 26 h (long term). Cell death was analyzed via flow cytometry using Annexin V and a viability nuclear dye (7-AAD) (Figure 1a and b). There were only a few dead cells after a 2 h incubation with H2O2 (7.5±0.52%), whereas long-term application of H2O2 (26 h) led to an immense increase in dead cells (86.5±4.8%).

Long-term oxidative stress induces caspase-independent T-cell death in primary human T cells. (a) Primary human T cells (PBTs) were incubated with 100 μM H2O2 for the indicated time points. Cell death was quantified with Annexin V (y axis) and 7-AAD (x axis). The upper left quadrant shows the early apoptotic cells and the upper right quadrant shows the late apoptotic cells. (b) The graph shows a quantification of late apoptotic cells as shown in (a) (n=3; S.E.M.) (***P<0.001; NS: not significant). (c) PBTs were treated with the indicated concentrations of H2O2 for 26 h in the presence (black columns) or absence (white columns) of the pan-caspase inhibitor Z-VAD-fmk (20 mM). Cell death was analyzed using Annexin V/7-AAD staining as described in (a) and (b) (n=3; S.E.M.). (d) Jurkat T cells were treated with 1 μg/ml CD95 antibody in the presence or absence of Z-VAD-fmk. Cell death was assessed as described above
To determine the threshold for the induction of cell death upon oxidative stress, we exposed T cells to increasing levels of H2O2 for 26 h. Interestingly, only a small number of dead cells were found in samples with H2O2 concentrations lower than 12.5 μM H2O2 (e.g. 21±1.8% dead cells at 12.5 μM H2O2) (Figure 1c). On the contrary, substantially more cell death was induced in the presence of 50 μM (86.4±6.5% dead cells) or 100 μM H2O2 (92.6±3.1% dead cells) (Figure 1c). In parallel experiments, we scrutinized whether cell death induced during oxidative stress depended on caspase activation. However, the broad caspase inhibitor Z-VAD-fmk did not influence cell death of oxidatively stressed T cells (Figure 1c), although this inhibitor reduced CD95-triggered classical apoptosis in T cells (Figure 1d). These experiments clearly show that long-term oxidative stress induced a caspase-independent type of cell death in primary human T cells.
Long-term oxidative stress induces necrotic-like cell death in T cells
The type of cell death shown above could be of type I (apoptosis or apoptotic-like PCD), type II (autophagy) or type III (necrotic-like PCD or accidental necrosis). Type I can be distinguished from type II and type III by analyzing the morphology of the DNA/nuclei. Thus, to dissect the different types of cell death, we first analyzed the morphology of the DNA/nuclei. The morphology of stained nuclei was evaluated on the one hand by visual consideration, and on the other hand by an objective quantification of the area and bright detail intensity of the nuclei stain using multispectral imaging flow cytometry (MIFC).9 The nuclei of long-term oxidatively stressed T cells showed hardly any difference in their morphology compared with untreated T cells (Figure 2a and b). Apoptotic cells with condensed chromatin have a smaller area of nuclei staining and the chromatin exhibits increased bright detail intensity. The latter was shown in parallel experiments where classical apoptosis was induced by CD95 (Figure 2c). The analysis of nuclear area and bright detail intensity showed that CD95 triggering led to a condensation of chromatin in 38.3%. As expected, these changes in morphology of the nuclei could be inhibited with Z-VAD-fmk, which confirms that this degradation process depended on caspases (Figure 2c and d).

Long-term oxidative stress leads to a necrotic-like cell death. (a, b) PBTs were incubated without (a, upper panel) or with 50 μM (a, lower panel) H2O2. A total of 10 000 cells were acquired using an ImageStream. The representative pictures show the dark field or side scatter (SSC, cyan), nuclear stain (green) and bright field. The dot plots of the cell populations on the right-hand side of (a) display the calculation of the area of the nuclear fragments (y axis) and bright detail intensity (x axis) on the single-cell level. Cells with nuclear fragments display higher nuclear bright detail intensity and smaller nuclear fragments area. The nuclear fragmentation is shown as percent of cells (for details, see Materials and Methods). A quantification of three independent experiments is shown in (b). (c, d) Jurkat T cells were treated with CD95 in the presence or absence of Z-VAD-fmk. The nuclear fragmentation was assessed for 10 000 cells as in (a) and (b). (e) Primary human T cells were incubated without (i and iv) or with (ii, iii, v, vi) 50 μM H2O2 for 26 h. Thereafter, cell morphology was analyzed using transmission electron microscopy (TEM). The lower lane shows pictures with higher magnifications of the respective cells in the upper lane. The bar in the lower lane corresponds to 500 and 1000 nm in the upper lane. The middle panel (ii and v) shows cells with a swollen phenotype of mitochondria. Shown are representative pictures taken from two independent experiments (M, mitochondria; N, nuclei; G, Golgi apparatus)
To analyze whether oxidative stress induced autophagy, we performed transmission electron microscopy (TEM). Figure 2e shows representative TEM pictures of control cells or two stages of oxidatively stressed T cells (Figure 2e). TEM analysis did not provide any evidence for autophagosome accumulation in oxidatively stressed T cells, which ruled out the possibility of autophagy. Furthermore, there was no evidence of elevated chromatin condensation in either of the cells, which confirms the data obtained with MIFC.
T-cell death of type III is associated with mitochondrial swelling and disintegration.10, 11 The mitochondria of late-stage oxidatively stressed T cells indeed showed a swollen and disintegrated phenotype by TEM (Figure 3a). To quantify this mitochondrial enlargement, we measured the mitochondrial area in a high number of T cells using MIFC. Indeed, the total area of mitochondria increased by about 30% from 8.43±0.74 μm2 in control cells to 11.01±0.86 μm2 in oxidatively stressed cells (Figure 3b). Moreover, we analyzed the membrane potential (ΔΨm) of the mitochondria under long-term oxidative stress using tetramethylrhodamine (TMRE) (Figure 3c and d). The mitochondrial membrane potential decreased strongly when cells were exposed to concentrations of H2O2 above 50 μM. Again, caspase inhibition did not significantly influence reduction of the mitochondrial membrane potential in oxidatively stressed T cells. Thus, oxidative stress induced necrotic-like PCD or accidental necrosis.

Mitochondrial disintegration upon oxidative stress. (a) Transmission electron microscopic pictures of the mitochondria from the control or oxidatively stressed (50 μM H2O2) T cells. Shown are representative pictures taken from two independent experiments (M, mitochondria). (b) The area (in μm2) of the mitochondria was analyzed using MIFC and is shown for untreated (gray histogram) and hydrogen peroxide-treated cells (black histogram). The graph shows the mean area of the mitochondria from untreated or hydrogen peroxide-treated T cells (n=3; S.E.; *P<0.05). (c, d) The mitochondrial membrane potential (ΔΨm) of unstressed (left histograms) or H2O2-stressed cells was assessed using TMRE. A decrease in the TMRE fluorescence corresponds to a loss of the membrane potential of the mitochondria. A quantification of three independent experiments is shown in (c). The black bars represent T cells that were pre-treated with Z-VAD-fmk (n=3; S.E.M.). Note that experiments using DilC1(5) instead of TMRE gave essentially the same results (Supplementary Figure 1)
Cofilin translocates to the mitochondria under oxidative stress conditions
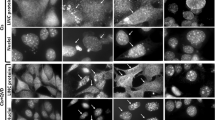
We have recently shown that cofilin is a major target of H2O2 in human T cells. Thus, pro-oxidants lead to the formation of an intramolecular disulfide bond between Cys-39 and Cys-80.4 Cofilin was reported to translocate to the mitochondria and mediate cell death of HL60 cells in response to staurosporine treatment by an as-yet unknown mechanism.12 As staurosporine increases intracellular ROS levels,13 and cofilin is oxidized under oxidative stress conditions,4 it was tempting to speculate that the oxidation of cofilin may be the key for its subsequent translocation into the mitochondria and induction of cell death under H2O2-induced oxidative stress conditions in primary human T cells. Indeed, cofilin colocalized with the mitochondria after treatment with 50 μM H2O2 as shown by confocal laser scan microscopy (Figure 4a). Moreover, cryo-immunogold electron microscopy revealed that cofilin could only be found inside the mitochondria of oxidatively stressed T cells, whereas it was at the boundaries of the mitochondria both in the absence and presence of oxidative stress (Figure 4b). To put the mitochondrial localization of cofilin into more quantitative terms, we used a logarithmic rescaling of the Pearson's correlation coefficient, that is a similarity score using MIFC,14, 15, 16, 17 for the respective T-cell populations (Figure 4c and d). In contrast to confocal laser scan or cryo-immunogold electron microscopy, this method allows an unbiased batch calculation of the cofilin localization in up to 10 000 cells. Again, a higher similarity score for cofilin and mitochondria was found in oxidatively stressed T cells compared with their unstressed counterparts (Figure 4c and d). Taken together, these data show that a fraction of cofilin translocated into the mitochondria under long-term oxidative stress conditions.

Cofilin translocates to the mitochondria upon oxidative stress. (a) Primary human T cells were incubated with (lower panel) or without (upper panel) 50 μM H2O2. Thereafter, cells were stained for cofilin (red) or mitochondria (MitoTracker, green) and analyzed via confocal laser scan microscopy. Merge displays the digital overlay of red and green fluorescence. The figure is representative of three independent experiments. (b) For cryo-immunogold electron microscopy, primary human T cells were either left untreated (i and iii) or treated with H2O2 (ii and iv) and subsequently fixed with 2% PFA for 10 min. Cells were stained with cofilin antiserum combined with protein A labeled with 15 nm gold particles. Shown are two example pictures taken from two independent experiments (M, mitochondria; N, nucleus). (c, d) The colocalization of cofilin and mitochondria was evaluated by the calculation of a similarity score of the two probes from untreated (gray histogram) and H2O2-treated (black lined histogram) PBT using MIFC. The histogram shows the distribution of the similarity within the whole-cell population as in conventional flow cytometry (up to 10 000 cells). A score of 1 indicates that the two probes are uncorrelated, whereas higher numbers indicate a higher degree of similarity. The mean similarity score of four independent experiments is shown in (d) (n=4; S.E.M.; *P<0.05)
Cofilin mutants that mimic oxidation translocate to the mitochondria and induce caspase- and PARP-independent cell death in primary human T cells
To analyze the role of cofilin oxidation for its mitochondrial translocation and the induction of cell death, we mutated the oxidation-sensitive cysteins of cofilin.4 According to previous data, a mutation can lead to either oxidation-mimicking mutants, if cysteins are exchanged by glycine,4 or oxidation-resistant mutants bearing alanine at the corresponding cysteins.18 To test this in parallel, we transfected wt-, G39- (Cys-39 to Gly-39) and A39-cofilin (Cys-39 to Ala-39) as eGFP-tagged versions in T cells. As expected, oxidative stress induced cell death in wt-cofilin-expressing T cells. However, G39-cofilin induced T-cell death even in the absence of oxidative stress (Figure 5a). The amount of dead G39-cofilin-expressing T cells could be increased by H2O2 to the same level as observed for H2O2-treated wt-cofilin-expressing T cells. Unlike G39-cofilin, A39-cofilin expression itself hardly induced any cell death. The number of dead T cells was even significantly reduced if oxidatively stressed T cells expressed A39-cofilin. These data suggest that a mutation to alanine interferes with the oxidation of cofilin, whereas a mutation to glycine functionally mimic oxidized cofilin.4 The substitution of the second cystein involved in disulfide bond formation (Cys-80) by glycine (G80-cofilin)4 was also able to induce PCD in human T cells, being even more efficient than G39-cofilin (Figures 5b and e). As observed for H2O2 treatment, the membrane potential of mitochondria decreased after G39-cofilin and G80-cofilin expression (Figure 5c). Interestingly, although these cofilin mutants induced disintegration of mitochondria and provoked Z-VAD-fmk-sensitive activation of caspase-3 (Figure 5d), they did not induce apoptosis. Rather, in line with the findings obtained upon long-term oxidative stress, the cell death induced by the expression of these cofilin mutants was resistant toward such caspase inhibition (Figure 5e).

Cofilin mutants translocate into the mitochondria and induce PCD in primary human T cells. (a) T cells expressing wt-, G39- or A39-cofilin were incubated in the absence (white bars) or presence (black bars) of H2O2. The percentages of dead cells were analyzed as described above (n=3; S.E.M.; #P<0.05 compared with wt-cofilin without H2O2; *P<0.05 compared with wt-cofilin with H2O2). (b) T cells expressing eGFP-tagged wt-cofilin, G39-cofilin or G80-cofilin were stained for Annexin V (y axis) and 7-AAD (x axis). The percentage of late apoptotic cells is depicted in the upper right corner of the dot plot. (c) The mitochondrial membrane potential was analyzed in wt-, G39- or G80-cofilin-expressing T cells using DilC1(5) as described (n=3; S.E.; *P<0.05). Note that TMRE staining yielded similar results (not shown). (d, e) Wt-cofilin, G39-cofilin or G80-cofilin was incubated with (black bars) or without (white bars) Z-VAD-fmk for 26 h. The activation of caspase-3 (d) as well as cell death (e) was analyzed using flow cytometry (n=3; S.E.M.). (f) Primary human T cells were incubated with the indicated amounts of H2O2 for 26 h in the presence (black bars) or absence (white bar) of 500 nM PJ-34. Cell death was assessed by flow cytometry using 7-AAD and Annexin V. The bars represent the mean of three independent experiments and S.E.M. (g) Cofilin constructs (wt-, G39- or G80-Cofilin) were transfected into primary human T cells. The T cells were then incubated without (white bars) or with 500 nM (black bars) or with 1000 nM (gray bars) of the PARP inhibitor PJ-34. The percentage of dead cells was determined as described above (n=3; S.E.M.)
Poly-(ADP-ribose) polymerase-1 (PARP) is an enzyme that was recently shown to mediate mitochondrial disintegration and cell death after oxidative stress.8, 19 Therefore, we tested whether PARP intervenes in the oxidized cofilin death pathway. However, although inhibition of PARP with PJ-34 partially reduced cell death induced by H2O2 (Figure 5f), PJ-34 did not influence cell death in T cells induced through the expression of G39- or G80-cofilin (Figure 5g). Thus, PARP does not seem to act downstream of oxidized cofilin. This information leads us to think that PARPs’ major influence on H2O2-triggered PCD probably occurs in a parallel way to the cofilin oxidation pathway.
From the morphological point of view, G39- and G80-cofilin behaved like endogenous or wt-cofilin upon H2O2 treatment of T cells. Thus, G39- and G80-cofilin predominantly colocalized with the mitochondria (Figure 6a). The similarity score of the cofilin constructs G39- and most notably G80-cofilin and mitochondria was increased compared with wt-cofilin (Figure 6b). Interestingly, mitochondrial size increased by 50% in G80-cofilin-expressing cells compared with wt-cofilin-expressing cells (Figure 6c). As observed for long-term oxidatively stressed T cells, the morphology of the nuclei was indistinguishable between wt-, G39- and G80-cofilin-expressing T cells (Figure 6a and d). These results show that the expression of cystein-to-glycine mutants of cofilin induced the same phenotype as observed for long-term H2O2 exposure and thus mimicked oxidative stress-induced necrotic-like PCD.

Oxidation-mimicking mutants of cofilin translocate into the mitochondria and induce programmed cell death. (a) Transfected T cells were stained for mitochondria (red) and analyzed by confocal laser scan microscopy. The eGFP-tagged cofilin is shown in green. The localization was analyzed for at least 500 cells per construct using MIFC. (b) The colocalization of mitochondria and cofilin was evaluated using the similarity score (MIFC analysis). The higher background colocalization originates from the high expression levels of the eGFP-tagged cofilin constructs as well as the tendency of eGFP to disperse throughout the cell. (c, d) The area and nuclear fragmentation of eGFP-positive cells was assessed as described above
Primary human T cells are of small size and eGFP, which was fused to the cofilin mutants, tends to disperse throughout the cells. The similarity score for wt-cofilin is therefore higher than that observed for endogenous cofilin. To rule out artificially high similarity scores, we compared the mitochondrial localization of wt-, G39- and G80-cofilin in COS cells using confocal laserscan microscopy (Supplementary Figure 2). We could confirm the results obtained for T cells with this high-resolution technique. Both G39-cofilin and G80-cofilin strongly localized in the mitochondria, whereas wt-cofilin was evenly distributed throughout the cell.
Taken together, the expression of cofilin with a single point mutation at either Cys-39 or Cys-80 was sufficient to induce necrotic-like PCD. The phenotype of T cells expressing G39- and G80-cofilin resembled the phenotype of long-term oxidatively stressed T cells.
Heat-shock cognate protein 70 associates with cofilin under oxidative stress conditions
To get mechanistic insights into how the mitochondrial localization of cofilin under long-term oxidative stress is regulated, we expressed FLAG-tagged wt-cofilin G39- or G80-cofilin in Jurkat T cells. Transfected cells were lysed and cDNA-expressed cofilin was immunoprecipitated by its FLAG-tag (Figure 7a and b). Immunoprecipitated proteins were analyzed by SDS-PAGE followed by mass spectrometry (Figure 7a). Wt- and G39-cofilin interacted equally with actin. This is in line with the finding that oxidation of cofilin did not inhibit actin binding.4 In contrast to wt-cofilin, a protein of 70 kDa, identified as heat-shock cognate protein 70 (HSC70), co-precipitated with G39-cofilin. Binding of HSC70 to G39- and G80-cofilin was confirmed by immunoprecipitation and western blot analysis of cofilin mutants (Figure 7b). A co-precipitation of HSC70 could be observed for both G39- and G80-cofilin.

Cofilin binds to HSC70 under oxidative stress conditions. (a) FLAG-tagged wt- or G39-cofilin was expressed in and then precipitated from Jurkat T cells. Lysates were subjected to PAGE and stained with Coomassie blue. The heavy (50 kDa) and light (ca. 25 kDa) chains of the precipitating antibody were found in each lane, including the control (untransfected Jurkat T cells). Bands that were found only in lane 2 (G39-cofilin) or lane 3 (wt-cofilin) were analyzed using mass spectrometry and identified as HSC70 (only present in lane 2) or actin (lanes 2 and 3). (b) Wt-, G39- or G80-cofilin was precipitated from Jurkat T cells. The corresponding western blot was stained for the FLAG-tag (lower part) or HSC70 (upper part). (c, d) PBTs were treated with 50 μM H2O2 for 7 or 24 h. Thereafter, cells were lysed and endogenous cofilin was immunoprecipitated. The precipitates were subjected to western blot analysis and stained for HSC70 (upper panel) or cofilin (lower panel). The graph in (d) shows a quantification of the HSC70 to cofilin ratios of three independent experiments (n=3; S.E.M.; *P<0.05). (e) The subcellular localization of HSC70 in long-term oxidatively stressed (lower part) or control T cells (upper part) was analyzed using confocal laser scan microscopy. The white color in the merge of the stressed cells shows the colocalization of cofilin and HSC70 with the mitochondria. The figure is representative of three experiments. (f) The colocalization of cofilin and mitochondria (upper graph), cofilin and HSC70 (lower graph) or HSC70 and mitochondria (central graph) was evaluated by the calculation of a similarity score of the corresponding probes (n=3; S.E.M.; *P<0.05)
As G39- and G80-cofilin represent oxidation-mimicking mutants,4 these results suggested that endogenous cofilin may bind to HSC70 under oxidative stress conditions. To elucidate this point, we incubated primary human T cells with 50 μM H2O2 for 7 and 24 h, respectively, and immunoprecipitated cofilin from lysates of these cells (Figure 5c). HSC70 indeed associated with cofilin under oxidative stress conditions. The association was most prominent under long-term oxidative stress conditions, that is, after 24 h treatment with H2O2. An association of cofilin with HSC70 could also be observed after 7 h. However, the co-immunoprecipitation was very weak compared with 24 h H2O2 treatment and the difference to untreated cells was not statistically significant (Figure 7d). The association of cofilin and HSC70 under oxidative stress could be important for the mitochondrial import of cofilin since HSC70 can chaperon mitochondrial import.20, 21 Thus, we finally examined the subcellular localization of HSC70 under long-term oxidative stress. Figure 7e shows that the incubation of T cells with 50 μM H2O2 for 26 h caused a colocalization of cofilin, HSC70 and mitochondrial staining. A calculation of the similarity scores showed a significant colocalization of cofilin with the mitochondria and HSC70 (Figure 7f). The colocalization between HSC70 and mitochondria increased upon H2O2 treatment, but did not reach a significant level, suggesting either a partial localization of the mitochondria and HSC70 or a localization of HSC70 at the outer membrane of the mitochondria. To analyze this point in more detail, we analyzed the subcellular localization of HSC70 in COS cells expressing wt-cofilin or the oxidation-mimicking mutant G39- or G80-cofilin using confocal laserscan microscopy (Supplementary Figure 3). Interestingly, in contrast to wt-cofilin-expressing COS cells, HSC70 enriches at the mitochondrial boundaries of G39- or G80-cofilin-expressing cells, which is most obvious if the mitochondria were in longitudinal orientation (Supplementary Figure 3). This localization of HSC70 is in line with the assumption that HSC70 chaperons the mitochondrial import of cofilin. Moreover, these data explain the small similarity score of HSC70 and mitochondria in oxidatively stressed T cells (Figure 7f).
Oxidized cofilin mediates cell death under oxidative stress conditions
To investigate whether cofilin and its mitochondrial localization were responsible for caspase-independent cell death triggered by long-term oxidative stress, we knocked down cofilin with siRNA (Figure 8a and b). These cofilin knockdown cells as well as control siRNA-transfected cells were then exposed to H2O2. In marked contrast to T cells transfected with control siRNA, only very few cofilin knockdown T cells died under these conditions (Figure 8c). These findings show that cofilin has an important role in inducing cell death under oxidative stress conditions. This, in addition, rules out the possibility of accidental necrosis after long-term oxidative stress conditions, as interfering with involved factors can interfere with oxidative stress-induced cell death.10

Knockdown of cofilin prevents necrotic-like PCD upon oxidative stress conditions. (a) T cells were treated with control or cofilin-specific siRNA. The expression of cofilin and actin was assessed in the lysates of these cells. Note that the mock-transfected cells were undistinguishable from control siRNA-treated cells (data not shown). (b) A quantification of three independent experiments shows that cofilin was efficiently knocked down by the siRNA (n=3; S.E.; **P<0.01). (c) The induction of cell death (Annexin V/7-AAD staining) was analyzed in control siRNA or cofilin-specific siRNA-treated cells that were either incubated with (black bars) or without (white bars) H2O2 for 26 h (n=3; S.E.; **P<0.01)
Discussion
The production of ROS influences the outcome of T-cell responses. Depending on concentration, exposure time and microenvironment, the effects of ROS on T cells can be very distinct. Thus, moderate H2O2 generation by T cells upon TCR triggering augments their immune responses,3, 22 whereas higher concentrations of exogenous H2O2 can either induce T-cell hyporesponsiveness4 or cell death (Hildeman et al5 and this work). This suppression of T cells is obviously harmful in cancer settings, but can be beneficial in autoimmune settings.2, 23, 24 Yet, only very little was known about the molecular switch, which serves as the checkpoint for the distinct consequences, that is, activation versus hyporesponsiveness or death of T cells. We show that oxidation and mitochondrial translocalization of cofilin leads to a point of no return by inducing necrotic-like PCD. Cofilin is resistant to low concentrations of H2O2, as they were observed on TCR triggering.3, 4, 22 However, high H2O2 concentrations can lead to cofilin oxidation and result in its mitochondrial localization and ultimately induces necrotic-like PCD and clearance of T cells from the site of inflammation.
Oxidative stress leads to the oxidation of several cellular proteins and lipids.8, 19, 25, 26 Yet, the specific importance of cofilin for the induction of PCD upon oxidative stress could be proven by (i) downregulation of cofilin with siRNA or (ii) expression of non-oxidizable cofilin (A39-cofilin). Both treatments protect primary human T cells from H2O2-induced necrotic-like PCD. However, it is worth noting that the expression of oxidation-mimicking cofilin mutants—G39- or G80-cofilin—is on its own sufficient to cause necrotic-like PCD and vice versa. One may speculate that the differences in the mutation to alanine or glycine might be caused by the fact that glycine has a low potential for the formation of α-helices and a relative high potential to form β-turns, and thus has a tendency to break the given secondary or ternary structures. In turn, alanine has a high tendency to stabilize α-helices and a low tendency to form β-turns. On the one hand, a mutation to glycine could thus introduce a β-turn, whereas alanine most likely does not. On the other hand, a mutation to alanine may inhibit disulfide bond formation without the induction of conformational changes, whereas a mutation to glycine may force and/or stabilize cofilin in its oxidation-mimicking conformation, which provokes its mitochondrial translocation.
The mitochondria have an important role in the induction or amplification of apoptotic, apoptotic-like and necrotic-like PCD. Mechanistically, loss of mitochondrial integrity is the key step that sentences the cells to death.27 Under normal conditions, antiapoptotic Bcl-2 family members, for example, Bcl-xL, maintain mitochondrial integrity. However, the translocation of pro-apoptotic members of the Bcl-2 family, for example, BAD and BAX, to the mitochondria favors the formation of permeability transition pores and eventually cell death. Obviously, the actin-binding protein cofilin can act on the mitochondria as a death-inducing agent that does not belong to the Bcl-2 family.
The differences between the short-term and long-term consequences of H2O2 can be due to either the amount of oxidized cofilin or the dependence of cofilin translocation into the mitochondria on an as-yet unknown factor. By far, most mitochondrial proteins are encoded in the nuclear genome and synthesized in the cytoplasm. The mitochondrial import then requires the mitochondrial translocase machinery. Cytoplasmic chaperones are crucially involved in the delivery of hydrophobic proteins. HSC70 has been identified as one chaperone that is important for the mitochondrial import.20 A similar mechanism seems to be responsible for the import of oxidized cofilin into the mitochondria, as HSC70 enriches at the boundaries of the mitochondria in oxidation-mimicking cofilin-expressing cells and only the cofilin forms mimicking oxidation efficiently co-immunoprecipitate HSC70.
So far, it was assumed that cell death induced upon mitochondrial localization of cofilin was due to apoptosis.12, 18, 28 However, our data clearly show that oxidized cofilin induces the phenotype of necrotic-like PCD, but not that of apoptosis. It is known that energy depletion causes a switch from apoptosis to necrotic-like PCD.29 As oxidation-mimicking cofilin mutants translocate into the mitochondria and induce necrotic-like PCD and not apoptosis, it is likely that oxidized cofilin interferes with mitochondrial functions and thus induces energy depletion. In addition, other proteins like PARP are oxidized under oxidative stress, which reinforces energy deprivation and forces T cells into necrotic-like PCD.30
Cell death induced through staurosporine treatment of human promyelocytic leukemia cells (HL60) and neuroblastoma cells (SH-SY5Y)12 or TGF-β treatment of human prostate carcinoma cells28 was also accompanied by translocation of cofilin to the mitochondria. Although the mechanisms of cofilin translocation into the mitochondria after staurosporine and TGF-β treatment are unknown, it is likely that this is due to a direct cofilin oxidation, as both TGF-β31 and staurosporine treatment13 can induce the production of intracellular ROS. At any rate, the dependence of PCD after long-term oxidative stress or TGF-β triggering on the mitochondrial localization of cofilin puts this protein in a central position in controlling immune responses.
Tumor necrosis factor (TNF) triggering can induce both types of PCD in fibroblasts, that is, apoptotic and necrotic.32, 33, 34, 35 Induction of necrotic-like PCD is most obvious in the presence of caspase inhibition.35 In human T cells, TNF, TNF-related apoptosis-inducing ligand (TRAIL) and even fibroblast-associated antigen (Fas) are able to induce necrotic-like PCD.36 This necrotic-like PCD is dependent on the expression of the kinase receptor interacting-protein (RIP). Interestingly, the action of RIP induces ROS production and, vice versa, ROS scavengers block the TNF-induced necrotic-like PCD in fibroblasts.33, 37 It is therefore tempting to speculate that oxidized cofilin may also facilitate the caspase-independent necrotic-like PCD induced by TNF, TRAIL or Fas in human T cells. The physiological necessity of such an alternative way of PCD becomes obvious by the fact that many viruses encode caspase inhibitors.38 If the organism would be completely dependent on caspases to clear viral infections, humans would be defenseless against viruses that are capable of producing caspase inhibitors.
Materials and Methods
Cell culture, reagents and antibodies
Human PBMCs were obtained by Ficoll–Hypaque (Linaris, Wertheim-Bettingen, Germany) density-gradient centrifugation of heparinized blood from healthy volunteers. T cells were purified with negative magnetic bead selection using the ‘Pan T-cell isolation Kit’ (Miltenyi Biotech, Bergisch Gladbach, Germany) as per the manufacturer's instructions. MitoTracker staining was performed in complete medium for 30 min at 37°C. Unbound MitoTracker was removed by extensive washing. Thereafter, the T cells were used in the experiments as indicated.
The mouse monoclonal anti-FLAG antibody (clone M2, 0.2 μg/ml) and cytochalasin D were from Sigma-Aldrich (Taufkirchen, Germany); cofilin antibodies were produced in our laboratory; HSC70 antiserum was from Santa Cruz (Heidelberg, Germany); the CD95 antibody was a kind gift of PH Krammer (DKFZ, Heidelberg, Germany); Z-VAD-fmk was from Promega (Mannheim, Germany); 7-AAD, Annexin V and anti-active caspase-3 were from BD Bioscience (Heidelberg, Germany); Hoechst 33342, MitoTracker Deep Red FM and DilC1(5), as well as TMRE were obtained from Invitrogen (Karlsruhe, Germany); and PJ-34 was purchased from Sigma.
Transfection procedures
For cDNA transfection of primary human T cells, the ‘Human T-Cell Nucleofector Kit’ (Amaxa Biosystems, Cologne, Germany) was used as per the manufacturer's instructions.
For siRNA experiments, Jurkat T cells were transfected via electroporation using the Bio-Rad GenePulser II (München, Germany). Cells (10 × 106 in 400 μl PBS containing Ca2+ and Mg2+) were mixed with up to 2 μg siRNA (non-targeting control siRNAs and cofilin siRNAs from Dharmacon, Lafayette, Colorado, USA) and electroporated at 230 V and 950 μF. Thereafter, they were transferred into 30 ml RPMI 1640 containing 10% FCS and incubated for 48 h in RPMI 1640 containing 10% FCS at 5% CO2 and 37°C. Then, cells were re-transfected with 2 μg siRNA and incubated at 5% CO2 and 37°C in RPMI 1640 containing 10% FCS for 72 h before analysis.
Immunoprecipitation
Immunoprecipitation was essentially carried out as described.39 Briefly, post-nuclear lysates were incubated overnight at 4°C with antibodies against the indicated targets or irrelevant control antibodies. Subsequently, pre-equilibrated protein-G sepharose was added, followed by an incubation for 1.5 h at 4°C. The samples were then washed five times and the precipitated proteins were solubilized in SDS sample buffer. The precipitated proteins were separated on SDS-PAGE and detected with specific antibodies using western blot analysis as indicated. Alternatively, the gels were stained with Coomassie blue, the bands excised and the corresponding proteins were identified using mass spectrometry at the EMBL core facility (Heidelberg, Germany).
Multispectral imaging flow cytometry
Data acquisition was performed with an ImageStream (Amnis, Seattle, WA, USA), which is a hybrid system combining a fluorescence microscope and a flow cytometer. Thus, it allows the acquisition of several thousand images per sample with a custom-designed × 40 objective (0.75 NA). Image data were analyzed using IDEAS 3.0 (Amnis), which analyzes thousands of images per sample on the single-cell level. As data mining is performed in an automated batch operation, the data are objective and unbiased.
For the analysis of nuclear fragmentation, a Hoechst33342-dependent image mask was created that covers the brightest 30% of the nuclear stain. This mask was used to calculate the area of the nuclear fragments. Moreover, the standard IDEAS feature ‘bright detail intensity’ was applied, which calculated the background-corrected intensity of bright spots with a radius of 3 pixels. Nuclear fragmentation leads to a reduction in the area of the bright nuclear stain and the bright detail intensity increases.9, 40
The mitochondrial area was calculated as the size of the morphology mask around the mitochondrial stain and the mitochondrial localization of cofilin or HSC70 was assessed using the IDEAS similarity feature, which is a log-transformed Pearson's correlation coefficient.14, 15, 16
Confocal laser scan microscopy
Confocal laser scan microscopic analyses were essentially performed as described.4 Briefly, cells were fixed with 2% paraformaldehyde for 10 min at room temperature and subsequently stained with the indicated antibodies. After staining, digitized confocal images were generated using a confocal laser-scanning microscope (Leica DMRBE with TCS NT, Bensheim, Germany) with an × 63 objective (1.4 NA).
Transmission electron microscopy
T cells were fixed with a mixture of 2% glutaraldehyde and 4% formaldehyde in 0.1 M PHEM buffer, pH 6.9 (60 mM PIPES, 25 mM HEPES, 2 mM MgCl2, 10 mM EGTA). After washing, the samples were post-fixed with 2% osmium tetroxide/1.5% potassium ferrocyanide for 1 h, washed, contrasted en bloc with uranyl acetate, dehydrated with an ascending series of ethanol and embedded in glycid ether 100-based resin. Ultrathin sections were cut with a Reichert ULTRACUT S ultramicrotome (Leica Microsystems, Wetzlar, Germany). After contrasting with uranyl acetate and lead citrate, they were viewed with an EM 10 CR electron microscope (Carl Zeiss NTS, Oberkochen, Germany).
The cofilin antibody did not detect its epitope if cells were fixed with higher concentrations of paraformaldehyde and glutaraldehyde. Therefore, the fixation conditions were changed for cryo-immunogold electron microscopy: cells were fixed with 2% formaldehyde in PHEM buffer, pH 6.9, for 10 min. Thereafter, the samples were quenched with 0.15 M glycine, embedded in 12% gelatin, infiltrated with 2.3 M sucrose, mounted, frozen and cryosectioned. The sections were picked up using a 1+1 mixture of 2.3 M sucrose in PBS and 2% methyl cellulose in water and transferred onto Pioloform/carbon-coated grids. Cryosections were blocked with 1% BSA in PBS for 10 min and incubated with anti-cofilin antibodies followed by protein A 15 nm gold for 30 min. After fixation with 1% glutaraldehyde, the sections were washed with water and contrasted. Note that the fuzziness of the cryo-immunogold electron microscopy pictures is owing to the mild fixation.
Analysis of life/dead and mitochondrial membrane potential using flow cytometry
The cells (2 × 105/200 μl) were incubated with or without the mitochondrial membrane potential dye DilC1(5) (25 nM) or TMRE (500 nM) for 15 min at 37°C. Two washing steps then removed the excessive dye. The cells were incubated in 200 μl RPMI 1640 containing 10% FCS for the indicated time points at 5% CO2 and 37°C in the presence or absence of H2O2 or 1 μg/ml CD95 antibodies. The cells were spun down, the supernatant aspirated, the cell pellet suspended in 50 μl 7-AAD and Annexin V-containing buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) and incubated for 20 min at room temperature. After extensive washing, the cells were analyzed with an LSRII (BD Bioscience).
Statistics
The statistical analysis was performed with GraphPad Prism version 4.00 (La Jolla, California, USA). The two groups were compared using t-test or paired t-test for matched observation.
Abbreviations
- ROS:
-
reactive oxygen species
- PCD:
-
programmed cell death
- TNF:
-
tumor necrosis factor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- RIP:
-
kinase receptor interacting protein
References
Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T . Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci USA 1996; 93: 13119–13124.
Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R et al. Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest 2007; 117: 3020–3028.
Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS . T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol 2004; 5: 818–827.
Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y . Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity 2008; 29: 404–413.
Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity 1999; 10: 735–744.
Kaminski M, Kiessling M, Suss D, Krammer PH, Gulow K . Novel role for mitochondria: protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol 2007; 27: 3625–3639.
Samstag Y, Eibert SM, Klemke M, Wabnitz GH . Actin cytoskeletal dynamics in T lymphocyte activation and migration. J Leukocyte Biol 2003; 73: 30–48.
Thoren FB, Romero AI, Hellstrand K . Oxygen radicals induce poly(ADP-ribose) polymerase-dependent cell death in cytotoxic lymphocytes. J Immunol 2006; 176: 7301–7307.
George TC, Basiji DA, Hall BE, Lynch DH, Ortyn WE, Perry DJ et al. Distinguishing modes of cell death using the ImageStream multispectral imaging flow cytometer. Cytometry A 2004; 59: 237–245.
Jaattela M, Tschopp J . Caspase-independent cell death in T lymphocytes. Nat Immunol 2003; 4: 416–423.
Golstein P, Kroemer G . Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 2007; 32: 37–43.
Chua BT, Volbracht C, Tan KO, Li R, Yu VC, Li P . Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat Cell Biol 2003; 5: 1083–1089.
Kruman I, Guo Q, Mattson MP . Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res 1998; 51: 293–308.
French AP, Mills S, Swarup R, Bennett MJ, Pridmore TP . Colocalization of fluorescent markers in confocal microscope images of plant cells. Nat Protocol 2008; 3: 619–628.
George TC, Fanning SL, Fitzgeral-Bocarsly P, Medeiros RB, Highfill S, Shimizu Y et al. Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J Immunol Methods 2006; 311: 117–129.
Beum PV, Lindorfer MA, Hall BE, George TC, Frost K, Morrissey PJ et al. Quantitative analysis of protein co-localization on B cells opsonized with rituximab and complement using the ImageStream multispectral imaging flow cytometer. J Immunol Methods 2006; 317: 90–99.
Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA . Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 1992; 103: 857–862.
Klamt F, Zdanov S, Levine RL, Pariser A, Zhang Y, Zhang B et al. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat Cell Biol 2009; Sep: 6.
Huang Q, Wu YT, Tan HL, Ong CN, Shen HM . A novel function of poly(ADP-ribose) polymerase-1 in modulation of autophagy and necrosis under oxidative stress. Cell Death Differ 2009; 16: 264–277.
Artigues A, Iriarte A, Martinez-Carrion M . Binding to chaperones allows import of a purified mitochondrial precursor into mitochondria. J Biol Chem 2002; 277: 25047–25055.
Artigues A, Crawford DL, Iriarte A, Martinez-Carrion M . Divergent Hsc70 binding properties of mitochondrial and cytosolic aspartate aminotransferase. Implications for their segregation to different cellular compartments. J Biol Chem 1998; 273: 33130–33134.
Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS . Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med 2002; 195: 59–70.
Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J, Holmdahl R . Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci USA 2004; 101: 12646–12651.
Olofsson P, Holmberg J, Tordsson J, Lu S, Akerstrom B, Holmdahl R . Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet 2003; 33: 25–32.
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002; 297: 259–263.
Cross JV, Templeton DJ . Thiol oxidation of cell signaling proteins: controlling an apoptotic equilibrium. J Cell Biochem 2004; 93: 104–111.
Krammer PH, Arnold R, Lavrik IN . Life and death in peripheral T cells. Nat Rev Immunol 2007; 7: 532–542.
Zhu B, Fukada K, Zhu H, Kyprianou N . Prohibitin and cofilin are intracellular effectors of transforming growth factor beta signaling in human prostate cancer cells. Cancer Res 2006; 66: 8640–8647.
Leist M, Single B, Naumann H, Fava E, Simon B, Kuhnle S et al. Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp Cell Res 1999; 249: 396–403.
Filipovic DM, Meng X, Reeves WB . Inhibition of PARP prevents oxidant-induced necrosis but not apoptosis in LLC-PK1 cells. Am J Physiol 1999; 277: F428–F436.
Thannickal VJ, Aldweib KD, Fanburg BL . Tyrosine phosphorylation regulates H2O2 production in lung fibroblasts stimulated by transforming growth factor beta1. J Biol Chem 1998; 273: 23611–23615.
Denecker G, Vercammen D, Steemans M, Vanden Berghe T, Brouckaert G, Van Loo G et al. Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ 2001; 8: 829–840.
Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem 2004; 279: 10822–10828.
Sytwu HK, Liblau RS, McDevitt HO . The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity 1996; 5: 17–30.
Li M, Beg AA . Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol 2000; 74: 7470–7477.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1: 489–495.
Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell 2002; 13: 978–988.
Best SM . Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol 2008; 62: 171–192.
Ambach A, Saunus J, Konstandin M, Wesselborg S, Meuer SC, Samstag Y . The serine phosphatases PP1 and PP2A associate with and activate the actin-binding protein cofilin in human T lymphocytes. Eur J Immunol 2000; 30: 3422–3431.
Henery S, George T, Hall B, Basiji D, Ortyn W, Morrissey P . Quantitative image based apoptotic index measurement using multispectral imaging flow cytometry: a comparison with standard photometric methods. Apoptosis 2008; 13: 1054–1063.
Acknowledgements
We are grateful to Hilmar Bading, Department of Neurobiology and Interdisciplinary Center for Neurosciences, University of Heidelberg, for providing the opportunity to carry out the electron microscopy work in his laboratory. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB405/A4 and DFG SA393/3-3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Wabnitz, G., Goursot, C., Jahraus, B. et al. Mitochondrial translocation of oxidized cofilin induces caspase-independent necrotic-like programmed cell death of T cells. Cell Death Dis 1, e58 (2010). https://doi.org/10.1038/cddis.2010.36
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2010.36
Keywords
This article is cited by
-
Deadly actin collapse by disulfidptosis
Nature Cell Biology (2023)
-
The multiple links between actin and mitochondria
Nature Reviews Molecular Cell Biology (2023)
-
Cofilin1 oxidation links oxidative distress to mitochondrial demise and neuronal cell death
Cell Death & Disease (2021)
-
Electroacupuncture Attenuates Ischemic Brain Injury and Cellular Apoptosis via Mitochondrial Translocation of Cofilin
Chinese Journal of Integrative Medicine (2021)
-
Phosphorylation of cofilin-1 by ERK confers HDAC inhibitor resistance in hepatocellular carcinoma cells via decreased ROS-mediated mitochondria injury
Oncogene (2017)

{kind=link}
{kind=link}
{kind=link}