
Abstract
Bacilli are ubiquitous low G+C environmental Gram-positive bacteria that produce a wide assortment of specialized small molecules. Although their natural product biosynthetic potential is high, robust molecular tools to support the heterologous expression of large biosynthetic gene clusters in Bacillus hosts are rare. Herein we adapt transformation-associated recombination (TAR) in yeast to design a single genomic capture and expression vector for antibiotic production in Bacillus subtilis. After validating this direct cloning “plug-and-play” approach with surfactin, we genetically interrogated amicoumacin biosynthetic gene cluster from the marine isolate Bacillus subtilis 1779. Its heterologous expression allowed us to explore an unusual maturation process involving the N-acyl-asparagine pro-drug intermediates preamicoumacins, which are hydrolyzed by the asparagine-specific peptidase into the active component amicoumacin A. This work represents the first direct cloning based heterologous expression of natural products in the model organism B. subtilis and paves the way to the development of future genome mining efforts in this genus.
Similar content being viewed by others
Introduction
Next generation sequencing and genome mining technologies have revolutionized the discovery of natural product chemicals and biosynthetic enzymes that help fuel the fields of biotechnology and biomedicine1. Based on a recent comprehensive survey of publically available bacterial genomes, three phyla account for the majority of natural product biosynthetic potential, namely Actinobacteria, Proteobacteria and Firmicutes2. While sophisticated molecular biology techniques have been firmly established to connect biosynthetic gene clusters to encoded natural product molecules in Actinobacteria and Proteobacteria through the use of model expression hosts such as Streptomyces coelicolor and Escherichia coli, Firmicutes, which contain the natural product rich genus Bacillus, are lacking comparable molecular tools to support the heterologous expression of large natural product pathways3,4. Here we report the design and implementation of a versatile vector to support the direct capture of Bacillus biosynthetic gene clusters from genomic DNA by transformation-associated recombination (TAR)5 in yeast and heterologous expression in the model host Bacillus subtilis.
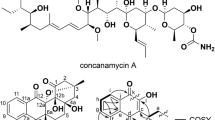
Bacillus subtilis is a low G+C, Gram-positive bacterium that has been commonly used for decades in genetic and biochemical studies of chromosome replication and bacterial sporulation6,7. This species is an attractive option for the heterologous production of natural products for three main reasons. First, the Bacillus genus produces a wide assortment of biologically active small molecules, including antibacterial non-ribosomal cyclic lipopeptides of the surfactin and gageotetrin families, polyketides such as macrolactin and bacillaene, antitumor polyketide-peptide hybrids like amicoumacin and ieodoglucomide and the discoipyrrole alkaloids (Fig. 1; Fig. S1)8,9,10,11,12. Second, B. subtilis has the capacity for natural genetic competence and subsequent homologous recombination, allowing the introduction of foreign DNA13,14. This feature offers a wide range of available genetic manipulation techniques to facilitate practical biosynthetic efforts of natural products. Indeed, the natural transformation system of B. subtilis is so effective that the 3.5-megabase genome of Synechocystis PCC6803 was successfully assembled into the B. subtilis 168 genome, which served as a cloning vector (also known as the Bacillus Genome (BGM) vector)14,15. This BGM cloning system has been applied to demonstrate the cloning of the entire mouse mitochondrion and rice chloroplast genomes16. And third, B. subtilis is non-pathogenic and is generally recognized as a safe production host that can satisfy safety requirements for the industrial productions of drug leads and enzymes17,18.

Chemical structure of amicoumacins (1–4) and surfactin.
Despite these technical advantages and that B. subtilis is routinely used for protein expression, there are few reports utilizing B. subtilis as an expression host system for the production of natural product small molecules. Most reports have used chromosomal transfer or cosmid library expression techniques and have focused on relatively small pathways of ribosomal and non-ribosomal peptide products19,20,21,22,23,24,25,26,27. However, the limitations of gene cluster cloning via clone libraries4 and chromosomal transfer hinder the efficient study of gene clusters from undomesticated producers or large pathways. Furthermore, a critical bottleneck in using B. subtilis as a heterologous host is the lack of autonomous plasmids to facilitate cloning, transfer and heterologous expression of large biosynthetic gene clusters3.
We recently developed a new genetic platform for the efficient capture of a silent 67-kb biosynthetic gene clusters directly from genomic DNA via TAR in yeast to support a “plug-and-play” approach to small molecule production28. To date we have captured and expressed high G+C Gram-positive actinomycete pathways for the marinopyrrole taromycin and enterocin28,29 antibiotics and the Gram-negative pseudoalteromonad pathway for the alterochromides30. Herein we adapted this platform to support the capture and expression of low G+C Gram-positive bacilli-based natural products. We validated the method with the prototype Bacillus lipopeptide surfactins and the hybrid polyketide-peptide amicoumacins, which exhibit broad bioactivities, including antibacterial, antifungal and antitumor activities12,31,32,33. This work represents a very useful approach to interrogate the function of biosynthetic gene clusters in Bacillus through heterologous biosynthesis.
Results
Design and validation of the pCAPB gene cluster capture vectors
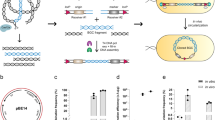
The gene cluster capture vector pCAP01 consists of three elements that allow direct capture and manipulation in yeast, maintenance and manipulation in Escherichia coli and chromosomal integration and expression of cloned pathways in actinomycetes28. To repurpose this vector for Bacillus expression, we first replaced the actinomycete elements with the Bacillus element from pBU434 to generate the yeast/E. coli shuttle-B. subtilis capture vector pCAPB1 in order to support conjugal transfer into various Bacillus species (Fig. S2). We evaluated this replication plasmid pCAPB1 with the prototype Bacillus lipopeptide surfactin, which is encoded on the 38-kb srf locus from B. subtilis 1779. Although we successfully captured the srf locus from genomic DNA via TAR and constructed the pCAPB1-srf vector (Fig. 2, Fig. S3), upon its transfer into five Bacillus host strains, the vector was not stable and did not allow for surfactin production.

Design and strategy of TAR-based cloning and expression.
(A) The procedure for TAR-based natural product heterologous expression in Bacillus involves three steps. In step 1, TAR in yeast involves homologous recombination between the linearized pathway specific capture vector and genomic DNA fragments to yield a circular construct that can form visible yeast colonies on selective media. In step 2, the cloned pathway can be manipulated using λ-Red recombination-mediated PCR targeting in E. coli. Finally, in step 3, through natural competence transformation, the cloned and manipulated pathway is integrated into the chromosome of Bacillus subtilis JH642 for natural products expression studies. (B) Physical map of the gene cluster capture vector pCAPB2 used in TAR direct cloning. The vector consists of three elements that allow direct cloning of pathways in yeast (blue), maintenance and manipulation in E. coli (yellow) and chromosomal integration and expression of cloned pathways in B. subtilis (red). The yeast element consists of ARSH4/CEN6 (replication origin) and TRP1 auxotrophic marker, while the E. coli and the Bacillus elements consist of DNA sequence for integration into the B. subtilis amyE gene, the lac repressor lacI,a spectinomycin resistance gene (specR) for Bacillus and an ampicillin resistance gene for E. coli (ampR). For the construction of a pathway specific capture vector, homology arms corresponding to both ends of the pathway are introduced into the capture arm cloning sites.
To overcome the instability issue of pCAPB1, we next designed pCAPB2 based on the amyE chromosomal integration plasmid pDR111, which is used in heterologous protein expression experiments in B. subtilis35,36. We incorporated the yeast elements from pCAP01 into a derivative of pDR111 to give the yeast/E. coli shuttle-B. subtilis chromosome integrative capture vector pCAPB2 (Fig. 2). This plasmid is maintained as a single copy in yeast cells to avoid unintended multiple recombination events during TAR, while it functions at multiple copies in E. coli to provide sufficient plasmid DNA materials for transfer to B. subtilis. The vector was designed to allow specific integration of cloned gene clusters into the chromosome of B. subtilis JH642 via double crossover recombination into the amyE gene. We validated the function of pCAPB2 by direct cloning of the srf gene cluster from B. subtilis 1779 to give the integration plasmid pCAPB2-srf (Fig. 2, Fig. S3). Upon its introduction into B. subtilis ROM77, in which the native srf locus was disrupted (JH642, srfAA::cat)37, we clearly detected surfactin production at wild-type levels by UPLC-MS (Fig. S4).
Identification and TAR capture of amicoumacin biosynthesis (ami) genes
With the successful completion of the proof-of-principle experiment to directly capture and express a Bacillus natural product pathway in B. subtilis, we next turned our attention to an uncharacterized Bacillus pathway to further showcase the proficiency of the pCAPB2 system. We selected to evaluate the amicoumacins, which are bioactive isocoumarin natural products that were first reported from Bacillus pumilus in 198131. Recent biochemical studies revealed that amicoumacin belongs to a new class of protein synthesis inhibitors that binds to the ribosome38,39. We re-isolated the amicoumacins from the Red Sea isolate B. subtilis 1779 following a bioassay-guided isolation procedure to give amicoumacins A-C (1-3) and O-methylamicoumacin B (4) (Fig. 1), based on high-resolution MS and NMR characterization. We suspect that some of the amicoumacin analogues may be byproducts generated from amicoumacin A (1) during the isolation and sample preparation processes. To capture and express the amicoumacin (ami) gene cluster, which we hypothesized to be encoded by a hybrid modular polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS), we sequenced the wild-type producer B. subtilis 1779 genome.
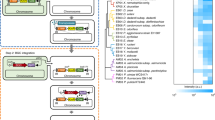
Sequence analysis revealed several assembly line biosynthetic gene clusters, including a contiguous region of 47.4 kb containing 16 open reading frames (ORFs) that we predicted were responsible for amicoumacin biosynthesis (Fig. 3A, Table S3). We designated these genes amiA-O. Inspection of this locus identified an NRPS-PKS hybrid protein encoded by amiI, two NRPSs encoded by amiA and amiJ and three PKS genes encoded by amiK-M. The predicted biosynthesis gene cluster consists of eight modules in total, as shown in Fig. 3, for the incorporation of three amino acids and five malonate residues. Based on the co-linearity rule of assembly line biosynthesis, we suspected that the predicted initiation module encoded by amiA synthesizes a fatty acyl-D-Asn residue reminiscent of lipopeptide natural products, suggesting that the product of the AmiA-M megasynthetase may be a derivative of lipoamicoumacins A–D that we previously reported40. We thus explored the possibility that the immediate product of the ami biosynthetic pathway may not be amicoumacin A but rather a lipidated precursor that may support a pro-drug-like activation mechanism (Fig. 3). The unusual dihydroisocoumarin core structure is likely formed by the terminating AmiJ–M megasynthetase proteins to generate a highly oxygenated polyketide chain that rearranges into the bicyclic dihydroisocoumarin moiety. Feeding experiments with 15N2-L-asparagine and 5,5,5-trifluoro-DL-leucine followed by MS analyses further supported the proposed amicoumacin A biosynthetic pathway (Table S4).

Proposed biosynthetic pathway of amicoumacins.
(A) Organization of the ami biosynthetic gene cluster in Bacillus subtilis 1779; (B) Proposed biosynthetic route to amicoumacins in B. subtilis 1779; (C) Postulated biosynthetic steps leading to hydroxymalonyl-ACP, which is a proposed PKS extender unit of the module 3. Domain abbreviations: A, adenylation; ACP, acyl carrier protein; AT, acyltransferase; C, condensation; E, epimerase; KR, ketoreductase; KS, ketosynthase; PCP, peptidyl carrier protein.
Owing to the projected unusual natural product activation mechanism, we initially interrogated the biosynthetic pathway of amicoumacins within the wild-type producer strain B. subtilis 1779. However, all of our attempts to disrupt target genes in the native strain were unsuccessful, as had been other attempts to previously analyze amicoumacin production41. Therefore, to study the amicoumacin biosynthesis pathway, we targeted the genomic region containing the 47.4-kb ami gene cluster for TAR and heterologous expression in either Bacillus subtilis or E. coli. We used 1-kb capture arms corresponding to the periphery of the ami locus to generate an ami pathway specific capture vector in pCAPB2. Saccharomyces cerevisiae VL6-48 was transformed with the linearized capture vector and genomic DNA fragments of B. subtilis 1779. Positive clones were identified by PCR and transferred to E. coli for propagation to give the heterologous expression construct pCAPB2-ami (Fig. 4, Fig. S5).

Physical maps of the TAR-cloned ami gene cluster and heterologous expression vectors.
The 47.4-kb genomic region containing the ami gene cluster (blue) was directly cloned in yeast, yielding pCAPB1-ami and pCAPB2-ami. The pCAPB1 backbone and unrelated gene orf1 on pCAPB1-ami (dotted arrows) were replaced with the ami specific capture vector pCAPE in E. coli cells via λ-red mediated recombination, generating ami gene cluster E. coli expression construct pCAPE-ami.
Heterologous expression of the amicoumacin biosynthesis genes
For the heterologous expression of the ami gene cluster, we introduced the integration plasmid pCAPB2-ami into the genome of B. subtilis JH642+sfp in which the phosphopantetheinyl transferase gene sfp has been added42. The resultant transformants successfully produced amicoumacins 1–4 at comparative levels to that in B. subtilis 1779, as revealed by UPLC-MS (Fig. 5A) and NMR analyses. We additionally expressed the ami cluster in E. coli BL21(DE3) via the construction of a different capture vector based on pETDuet-1, which also resulted in amicoumacin production, albeit at levels 100-fold less than that in the native B. subtilis 1779 strain or in the B. subtilis JH642 host (Fig. 5B, Fig. S6).

UPLC-MS analyses of heterologously produced amicoumacins.
(A) LCMS UV traces showing the relative production of amicoumacins (1–6) in the native B. subtilis 1779 and B. subtilis JH642+sfp carrying heterologous expression constructs pCAPB2-ami, pCAPB2-ami (ΔamiA), pCAPB2-ami (ΔamiB) and pCAPB2 (blank) with UV monitoring at 314 nm. (B) LCMS extracted ion chromatogram traces of amicoumacins (1-6) produced by E. coli BL21(DE3) carrying pCAPE-ami (0, 0.5 mM IPTG) and the empty vector pCAPE. (C) LCMS extracted ion chromatogram traces of compounds (1-5) showing the in vivo conversion of preamicoumacin A (5, “standard”) to amicoumacins (1–4) after 4 and 12 hours incubations in B. subtilis JH642+amiB.
The heterologous production of the amicoumacins in the B. subtilis and E. coli hosts provided unequivocal evidence that the ami locus encodes amicoumacin biosynthesis. With these systems in hand, we further interrogated the function of amiA by λ-Red recombination-mediated PCR targeting in E. coli BW2511343. Restriction mapping of the plasmid propagated in E. coli confirmed that the amiA gene was successfully replaced by a gene encoding apramycin resistance to yield pCAPB2-ami (ΔamiA) (Fig. S5). This construct was integrated into the chromosome of the B. subtilis host, whereupon we observed that amicoumacin production was now lost (Fig. 5A).
Pro-drug mechanism of amicoumacin activation
With the successful heterologous expression of the amicoumacin biosynthetic gene cluster and the ability to readily inactivate individual ami genes, we next explored the molecular and functional relationship between the various structures and the possibility of a pro-drug-like activation strategy. To this end, we first examined amiB, which codes for a D-Asn peptidase homologous to XcnG and ClbP that convert inactive precursors into the antibiotics xenocoumacin and colibactin, respectively44,45,46. To evaluate if AmiB is similarly involved in activating lipoamicoumacin-like precursors to form mature amicoumacin antibiotics, we mutated the amiB gene in the pCAPB2-ami plasmid (Fig. S5) and expressed pCAPB2-ami (ΔamiB) in the B. subtilis host. The mutant strain lacking amiB was then analyzed by UPLC-MS analysis (Fig. 5A). Indeed, as predicted, new shunt products were produced instead of amicoumacins in the amiB deletion mutant. These products were isolated and characterized as preamicoumacins A–B (5–6) (Fig. 3B) by comprehensive NMR, MS and Marfey analyses (Table S5, Fig. S7–S8 and Supplemental Experimental Procedures). Preamicoumacin A resembles lipoamicoumacin A and specifically differs from amicoumacin A by tailoring the amine group at C-10′ with N-acyl Asn as predicted bioinformatically. The configuration of the N-acyl Asn residue was assigned as D on the basis of the advanced Marfey’s methods47,48. Both 5 and 6 represent derivatives of amicoumacin A extended at the N terminus by D-Asn carrying two different acyl chains (Fig. 3B).
With the structures of preamicoumacins A–B in hand, we were able to investigate the biochemical function of the AmiB, which is predicted to be a membrane-associated peptidase. To directly observe the cleavage of preamicoumacins to amicoumacin A, we heterologously expressed AmiB in B. subtilis JH642 for in vivo tests of proteolytic activity against preamicoumacins. Addition of exogenous 5 to B. subtilis carrying the gene amiB resulted in its conversion to 1 (Fig. 5C). While amicoumacin A (1) is active against Staphylococcus aureus (UST950701-005) with an MIC of 5.0 µg mL−1, derivatives 5 and 6 were inactive (Table S6). These results support the biosynthetic scenario whereupon inactive preamicoumacin precursors are first synthesized and then converted to the active component amicoumacin A.
Discussion
Connecting genes to molecules with the help of efficient heterologous expression techniques is beginning to fundamentally change the natural product discovery paradigm. Herein we add the Bacillus antimicrobial compounds surfactin and amicoumacins to the small yet growing list of TAR captured and heterologously expressed microbial compounds28,29,30,49,50, thereby opening up the metabolically rich Bacillus genus to future natural product discovery efforts. In the present study, the TAR-directed capturing of the amicoumacin biosynthesis gene cluster from a marine B. subtilis isolate allowed for its heterologous expression and biosynthetic interrogation in a host B. subtilis strain, which represents a common genetic procedure practiced in other bacterial systems but not before with Bacillus. Our mutational work allowed us to establish an antibiotic “pro-drug” activation pathway in which newly discovered preamicoumacins are converted by the AmiB peptidase into the biologically active isocoumarin antibiotic in a process resembling xenocoumacin and colibactin processing in Gram-negative bacteria44,46 but not before observed in Gram-negative bacteria. This study was greatly facilitated with the pCAPB2 expression system that should similarly support the discovery and characterization of new chemical entities and enzymatic processes in other bacilli, which is an active pursuit of our laboratories.
Methods
Strains, fermentation and isolation of amicoumacin compounds
All strains, plasmids and oligonucleotides used in this study are listed in Table S1–S2. Bacillus subtilis 1779 was isolated from seawater collected from the Red Sea during a 2010 research cruise. Its crude extract showed strong antibacterial activity against S. aureus UST950701-005. Detailed culture conditions and isolation procedures of amicoumacins A–C and O-methylamicoumacin B (1–4) are described in the Supplemental Experimental Procedures. For identification of preamicoumacins A–B, B. subtilis JH642+sfp that carried pCABP1-ami (ΔamiB) was cultured in LB media for 24 hours, extracted with ethyl acetate and isolated by preparative RP-HPLC (60–100% MeCN in 0.1% trifluoroacetic acid, 40 min gradient) to obtain pure compounds. Analytical details of the preamicoumacins are provided in the Supplemental Information.
Genome sequencing, annotation and bioinformatics analysis of the ami gene cluster
The draft genome of B. subtilis 1779 was sequenced on an Illumina Hiseq2000 to generate 490-fold coverage of the 4.25 Mb genome. A total of 10.5 million pairs of Illumina reads were obtained from a 200 bp paired-end library. Genome assembly was performed with Velvet 1.0.15 with the following custom parameters: hash-length = 55 and coverage cutoff = 30. The draft genome sequences were deposited in GenBank as accession number SRS606572. The bioinformatics program antiSMASH (http://antismash.secondarymetabolites.org/)51 was initially used to analyze the whole draft genome sequence. The sequence of the orphan 47.4-kb NRPS/PKS hybrid gene cluster amiA-O encoded on contig-29 (95,452–140,246 nt) was further predicted and annotated using Pfam analyses (http://pfam.sanger.ac.uk/) and protein-protein BLAST (http://blast.ncbi.nlm.nih.gov). NRPS A domain specificities were analyzed using online program NRPSpredictor2 (http://nrps.informatik.uni-tuebingen.de/Controller?cmd=SubmitJob)51,52.
Construction of the gene cluster capture vectors pCAPB1 and pCAPB2
Our initial attempts of replication plasmid pCAPB1 generated from our previous capture vector for Streptomyces pCAP01 by replacing Streptomyces element with Bacillus element from plasmid pBU4 was not successful for heterologous expression in B. subtilis. To generate the integration vector pCAPB2, the yeast element consisting of ARSH4/CEN6 (replication origin) and TRP1 auxotrophic marker from pCAP01, the E. coli and the Bacillus elements consisting of DNA sequence for integration into the B. subtilis amyE gene, the lac repressor lacI and an IPTG-inducible promoter, a spectinomycin resistance gene (specR) for Bacillus and an ampicillin resistance gene for E. coli (ampR) from the pDR111 were assembled in E. coli Top10. For capture vector of heterologous expression in E. coli BL21 (DE3), the phosphopantetheine transferase (PPTase) gene sfp was inserted into MCS2 of pETDuet-1 to generate capture vector pCAPE. Detailed information is provided in Supplemental Experimental Procedures.
Direct cloning of the ami gene cluster using TAR
Producer strain B. subtilis 1779 was grown in LB liquid medium overnight and genomic DNA was isolated from stationary phase cells. Approximately, 20 μg of genomic DNA were digested with 400 U of ScaI or SpeI, which did not cut the ami or srf gene clusters, respectively, in an overnight reaction at 37°C. The ami pathway-specific capture vector was constructed by introducing two PCR-amplified 1-kb homology arms corresponding to upstream and downstream regions of the ami gene cluster (orf1 and orf3) into capture vector pCAPB1 and pCAPB2 (Fig. S2). To capture the ami gene cluster, spheroplast cells of S. cerevisiae VL6-48 were transformed with the linearized ami pathway-specific capture vector and enzymatically fragmented genomic DNA together. Desired transformants with the captured ami gene cluster were selected on synthetic tryptophan dropout agar and identified by PCR. Direct cloning of the ami cluster was confirmed by restriction mapping to give pCAPB1-ami and pCAPB2-ami. The surfactin gene cluster srf was similarly captured following the same protocol to give pCAPB1-srf and pCAPB2-srf. For the ami gene cluter expression in E. coli, competent cells of E. coli BW25113 carrying pIJ790 and pCAPB1-ami were transformed with the linearized capture vector pCAPE to generate pCAPE-ami via λ-Red mediated recombination. The pCAPB1-ami, pCAPB2-ami and pCAPE-ami constructs were obtained and confirmed by restriction mapping after stable propagation through E. coli (Fig. 4, Fig. S5). More detailed information is provided in Supplemental Experimental Procedures.
Heterologous expression of the ami gene cluster
The construct pCAPB2-ami and its derivatives, which have 1.0-kb homology regions corresponding to the upstream and downstream regions of the gene amyE, were transferred to strain B. subtilis JH642+sfp by natural competence transformation. Spectinomycin-resistant and PCR positive clones were routinely grown in LB broth containing spectinomycin (100 μg mL−1) at 30°C overnight. A portion (1.0 mL) of the preculture was inoculated into 100 mL of LB broth and grown for 1 d at 30°C in a 250-mL flask with rotary shaking. For heterologous expression in E. coli, construct pCAPE-ami was introduced into BL21 (DE3) cells via electroporation. The positive clones were inoculated and confirmed by restriction mapping and then similarly cultured. The EtOAc extracts from the culture broth were analyzed by reversed-phase UPLC-MS. Detailed information, including the analytical conditions for UPLC-MS, genetic manipulation of the genes amiA and amiB, sample preparation for UPLC-MS analysis and antimicrobial bioassay, are described in Supplemental Information.
References
Bachmann, B. O., Van Lanen, S. G. & Baltz, R. H. Microbial genome mining for accelerated natural products discovery: Is a renaissance in the making? J. Ind. Microbiol. Biotechnol. 41, 175–184 (2014).
Cimermancic, P. et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158, 412–421 (2014).
Zhang, H. R., Boghigian, B. A., Armando, J. & Pfeifer, B. A. Methods and options for the heterologous production of complex natural products. Nat. Prod. Rep. 28, 125–151 (2011).
Ongley, S. E., Bian, X. Y., Neilan, B. A. & Muller, R. Recent advances in the heterologous expression of microbial natural product biosynthetic pathways. Nat. Prod. Rep. 30, 1121–1138 (2013).
Kouprina, N. & Larionov, V. Selective isolation of genomic loci from complex genomes by transformation-associated recombination cloning in the yeast Saccharomyces cerevisiae. Nat. Protoc. 3, 371–377 (2008).
Moszer, I. et al. Subtilist: The reference database for the Bacillus subtilis genome. Nucleic Acids Res. 30, 62–65 (2002).
Kunst, F. et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249–256 (1997).
Tareq, F. S. et al. Gageotetrins A-C, noncytotoxic antimicrobial linear lipopeptides from a marine bacterium Bacillus subtilis. Org. Lett. 16, 928–931 (2014).
Tareq, F. S. et al. Ieodoglucomides A and B from a marine-derived bacterium Bacillus licheniformis. Org. Lett. 14, 1464–1467 (2012).
Stein, T. Bacillus subtilis antibiotics: Structures, syntheses and specific functions. Mol. Microbiol. 56, 845–857 (2005).
Hu, Y. C. et al. Discoipyrroles A-D: Isolation, structure determination and synthesis of potent migration inhibitors from Bacillus hunanensis. J. Am. Chem. Soc. 135, 13387–13392 (2013).
Hamdache, A., Lamarti, A., Aleu, J. & Collado, I. G. Non-peptide metabolites from the genus Bacillus. J. Nat. Prod. 74, 893–899 (2011).
Dubnau, D. Genetic competence in Bacillus subtilis. Microbiol. Rev. 55, 395–424 (1991).
Itaya, M., Tsuge, K., Koizumi, M. & Fujita, K. Combining two genomes in one cell: Stable cloning of the Synechocystis PCC6803 genome in the Bacillus subtilis 168 genome. Proc. Natl. Acad. Sci. USA 102, 15971–15976 (2005).
Itaya, M. et al. Efficient cloning and engineering of giant DNAs in a novel Bacillus subtilis genome vector. J. Biochem. 128, 869–875 (2000).
Itaya, M., Fujita, K., Kuroki, A. & Tsuge, K. Bottom-up genome assembly using the Bacillus subtilis genome vector. Nat. Methods. 5, 41–43 (2008).
de Boer, A. S. & Diderichsen, B. On the safety of Bacillus subtilis and B. amyloliquefaciens - a review. Appl. Microbiol. Biotechnol. 36, 1–4 (1991).
Leuschner, R. G. K. et al. Qualified presumption of safety (QPS): A generic risk assessment approach for biological agents notified to the European food safety authority (EFSA). Trends. Food. Sci. Tech. 21, 425–435 (2010).
Borisova, S. A. et al. Biosynthesis of rhizocticins, antifungal phosphonate oligopeptides produced by Bacillus subtilis ATCC6633. Chem. Biol. 17, 28–37 (2010).
Choi, S. K. et al. Identification of a polymyxin synthetase gene cluster of Paenibacillus polymyxa and heterologous expression of the gene in Bacillus subtilis. J. Bacteriol. 191, 3350–3358 (2009).
Eppelmann, K., Doekel, S. & Marahiel, M. A. Engineered biosynthesis of the peptide antibiotic bacitracin in the surrogate host Bacillus subtilis. J. Biol. Chem. 276, 34824–34831 (2001).
Herzner, A. M. et al. Expression of the lantibiotic mersacidin in Bacillus amyloliquefaciens FZB42. PLoS One 6, e22389. (2011).
Liu, W. & Hansen, J. N. Conversion of Bacillus subtilis 168 to a subtilin producer by competence transformation. J. Bacteriol. 173, 7387–7390 (1991).
Tsuge, K. et al. Horizontal transfer of iturin a operon, itu, to Bacillus subtilis 168 and conversion into an iturin A producer. Antimicrob. Agents Chemother. 49, 4641–4648 (2005).
Yuksel, S. & Hansen, J. N. Transfer of nisin gene cluster from Lactococcus lactis ATCC 11454 into the chromosome of Bacillus subtilis 168. Appl. Microbiol. Biotechnol. 74, 640–649 (2007).
Zobel, S., Kumpfmuller, J., Sussmuth, R. D. & Schweder, T. Bacillus subtilis as heterologous host for the secretory production of the non-ribosomal cyclodepsipeptide enniatin. Appl. Microbiol. Biotechnol. 99, 681–691 (2015).
Tang, Y. et al. Heterologous expression of an orphan nrps gene cluster from Paenibacillus larvae in Escherichia coli revealed production of sevadicin. J. Biotechnol. 194, 112–114 (2015).
Yamanaka, K. et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin a. Proc. Natl. Acad. Sci. USA 111, 1957–1962 (2014).
Bonet, B. et al. Direct capture and heterologous expression of Salinispora natural product genes for the biosynthesis of enterocin. J. Nat. Prod. 10.1021/np500664q. (2014).
Ross, A. C., Gulland, L. E., Dorrestein, P. C. & Moore, B. S. Targeted capture and heterologous expression of the Pseudoalteromonas alterochromide gene cluster in Escherichia coli represents a promising natural product exploratory platform. ACS Synth. Biol., 10.1021/sb500280q. (2014)
Itoh, J. et al. Amicoumacin-A, a new antibiotic with strong antiinflammatory and antiulcer activity. J Antibiot. (Tokyo) 34, 611–613 (1981).
Peypoux, F., Bonmatin, J. M. & Wallach, J. Recent trends in the biochemistry of surfactin. Appl. Microbiol. Biotechnol. 51, 553–563 (1999).
Singh, P. & Cameotra, S. S. Potential applications of microbial surfactants in biomedical sciences. Trends Biotechnol. 22, 142–146 (2004).
Bourgouin, C., Delecluse, A., Delatorre, F. & Szulmajster, J. Transfer of the toxin protein genes of Bacillus sphaericus into Bacillus thuringiensis subsp israelensis and their expression. Appl. Environ. Microbiol. 56, 340–344 (1990).
van Ooij, C. & Losick, R. Subcellular localization of a small sporulation protein in Bacillus subtilis. J. Bacteriol. 185, 1391–1398 (2003).
Wagner, J. K., Marquis, K. A. & Rudner, D. Z. SirA enforces diploidy by inhibiting the replication initiator DNAa during spore formation in Bacillus subtilis. Mol. Microbiol. 73, 963–974 (2009).
Solomon, J. M., Magnuson, R., Srivastava, A. & Grossman, A. D. Convergent sensing pathways mediate response to two extracellular competence factors in Bacillus subtilis. Genes. Dev. 9, 547–558 (1995).
Lama, A. et al. Response of methicillin-resistant Staphylococcus aureus to amicoumacin A. PLoS One. 7, e34037 (2012).
Polikanov, Y. S. et al. Amicoumacin A inhibits translation by stabilizing mRNA interaction with the ribosome. Mol. Cell 56, 531–540 (2014).
Li, Y. X. et al. Five new amicoumacins isolated from a marine-derived bacterium Bacillus subtilis. Mar. Drugs 10, 319–328 (2012).
Pinchuk, I. V. et al. Amicoumacin antibiotic production and genetic diversity of Bacillus subtilis strains isolated from different habitats. Res. Microbiol. 153, 269–276 (2002).
Nakano, M. M., Marahiel, M. A. & Zuber, P. Identification of a genetic-locus required for biosynthesis of the lipopeptide antibiotic surfactin in Bacillus subtilis. J. Bacteriol. 170, 5662–5668 (1988).
Gust, B. et al. PCR-targeted streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. USA 100, 1541–1546 (2003).
Bian, X. Y. et al. In vivo evidence for a prodrug activation mechanism during colibactin maturation. Chembiochem 14, 1194–1197 (2013).
Reimer, D. & Bode, H. B. A natural prodrug activation mechanism in the biosynthesis of nonribosomal peptides. Nat. Prod. Rep. 31, 154–159 (2014).
Reimer, D. et al. A natural prodrug activation mechanism in nonribosomal peptide synthesis. Nat. Chem. Biol. 7, 888–890 (2011).
Fujii, K. et al. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Combination of Marfey's method with mass spectrometry and its practical application. Anal. Chem. 69, 5146–5151 (1997).
Fujii, K. et al. Further application of advanced Marfey's method for determination of absolute configuration of primary amino compound. Tetrahedron Lett. 39, 2579–2582 (1998).
Kallifidas, D. & Brady, S. F. Reassembly of functionally intact environmental DNA-derived biosynthetic gene clusters. Methods Enzymol. 517, 225–239 (2012).
Kim, J. H. et al. Cloning large natural product gene clusters from the environment: Piecing environmental DNA gene clusters back together with TAR. Biopolymers 93, 833–844 (2010).
Blin, K. et al. Antismash 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, W204–W212 (2013).
Rausch, C. et al. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMS). Nucleic Acids Res. 33, 5799–5808 (2005).
Acknowledgements
This study was generously supported by a grant (DY125-15-T-02) from China Ocean Mineral Resources Research and Development Association, award SA-C0040/UK-C0016 from the King Abdullah University of Science and Technology to P.Y.Q. and funding from the NIH (R01-GM085770) to B.S.M.
Author information
Authors and Affiliations
Contributions
Y.L., K.Y., R.K., B.S.M. and P.Q. designed the experiments, Y.L., Z.L. and W.Z. executed the experiments, Y.L., Z.L., K.Y., Y.X., W.Z., H.V., R.K., B.S.M. and P.Q. analyzed the data and Y.L., B.S.M. and P.Q. wrote the manuscript with input from all authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Directed natural product biosynthesis gene cluster capture and expression in the model bacterium Bacillus subtilis
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, Y., Li, Z., Yamanaka, K. et al. Directed natural product biosynthesis gene cluster capture and expression in the model bacterium Bacillus subtilis. Sci Rep 5, 9383 (2015). https://doi.org/10.1038/srep09383
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09383
This article is cited by
-
Molecular basis of antibiotic self-resistance in a bee larvae pathogen
Nature Communications (2022)
-
Novel amylomacins from seaweed-associated Bacillus amyloliquefaciens as prospective antimicrobial leads attenuating resistant bacteria
World Journal of Microbiology and Biotechnology (2021)
-
Progress in the Chemistry of Amino-Acid Derivatives of Isocoumarins and 3,4-Dihydroisocoumarins
Chemistry of Natural Compounds (2021)
-
Bacteria as genetically programmable producers of bioactive natural products
Nature Reviews Chemistry (2020)
-
Strategies for Discovering New Antibiotics from Bacteria in the Post-Genomic Era
Current Microbiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
