
Abstract
To find metastatic recurrence biomarkers of triple-negative breast cancer (TNBC) treated by neoadjuvant chemotherapy and anti-EGFR antibodies (NAT), we evaluated tumor genomic, transcriptomic, and immune features, using MSK-IMPACT assay, gene arrays, Nanostring technology, and TIL assessment on H&E. Six patients experienced a rapid fatal recurrence (RR) and other 6 had later non-fatal recurrences (LR). Before NAT, RR had low expression of 6 MHC class I and 13 MHC class II genes but were enriched in upregulated genes involved in the cell cycle-related pathways. Their TIL number before NAT in RR was very low (<5%) and did not increase after treatment. In post-NAT residual tumors, RR cases showed high expression of SOX2 and CXCR4. Our results indicate that high expression of cell cycle genes, combined with cold immunological phenotype, may predict strong TNBC resistance to NAT and rapid progression after it. This biomarker combination is worth validation in larger studies.
Similar content being viewed by others


Introduction
Triple-negative breast cancer (TNBC) is characterized by the lack of expression of estrogen receptor (ER) and progesterone receptor (PR) and the absence of amplification of ERBB2. It encompasses a large spectrum of breast malignancies and is highly heterogeneous in terms of histology, molecular features, and clinical behavior1. Although some of these tumors are characterized by low histological grade and good long-term prognosis after local treatment, the majority present with a high grade and aggressive nature reflected by an early peak of recurrence, fatal distant metastases, and the 5-year overall survival (OS) rate lower than the other subtypes of breast cancer2,3. Therefore, finding reliable predictors of life-threatening recurrences is an unmet need for TNBC.
Most TNBC recurrences develop within 5 years after the diagnosis4, and it is accepted that the risk of recurrence is higher in TNBC patients with a big residual tumor (RT) after neoadjuvant treatment (NAT)5. Balko and collaborators have elegantly shown that post-NAT TNBC RTs carry numerous genomic alterations and that 90% of these tumors have at least one actionable molecular lesion6. This study posited that molecular alterations found in the post-NAT residual disease may be the same present in micrometastases that will cause disease recurrence.
Beyond genomic anomalies, recent studies have highlighted the importance of TNBC immunological subtype both for response to NAT and for further clinical evolution. TNBCs rich in tumor-infiltrating lymphocytes (TILs), either before or after NAT, have better outcomes than those poor in TILs7,8. Besides TIL quantity, gene expression profiling has revealed the existence of several prognostically relevant TNBC subtypes, with different immune microenvironment composition and immune response within the breast tumor tissue9,10.
However, none of those subtypes or other gene signatures that emerged as recurrence predictors in TNBC is sufficiently validated to enter the clinics11,12,13,14,15,16,17. In addition, no biological marker has been proposed to identify TNBCs that will progress shortly after NAT. Tumor relapses occurring within the first year after NAT are considered manifestations of primary chemoresistance and their long-term favorable outcome is exceedingly rare. In those tumors, chemotherapy can even stimulate metastatic progression by selecting the cells with high stemness and proliferation or by modifying the tumor microenvironment to favorize the survival and multiplication of metastatic tumor cells18,19,20,21. Therefore, the patients carrying such tumors would benefit from early identification, ideally before NAT, and be offered more advanced therapy approaches than the standard anthracycline-taxane regimens.
On the other hand, real-life observations in the clinic confirm that certain TNBC patients will never experience recurrence, even left with a big RT and no adjuvant chemotherapy after NAT. If readily identified at the post-NAT surgery, this patient population could be included in clinical trials testing de-escalation of adjuvant treatments. However, studies showing how to recognize either the TNBCs which will rapidly progress after NAT or the ones which will remain under long-term control despite big post-NAT residual tumors, are lacking.
Here we report a comprehensive genomic and transcriptomic analysis, integrated with lymphocyte infiltration, of pre-treatment and post-NAT samples from TNBC patients undergoing chemotherapy and EGFR blockade22,23.
Results
Patient population
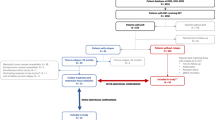
We analyzed a total of 62 patients, 12 of them experiencing metastatic recurrences. Six patients recurred within a year after surgery (group A) and rapidly deceased from metastatic disease. Six patients had later recurrences, mostly without a fatal outcome (in 1–5 years after surgery, group B) (Table 1 and Supplementary Fig. 1). Since we were interested in comparing molecular characteristics of the recurring tumors versus those of non-recurring tumors of various sizes, we further classified the non-recurring tumors into 4 groups: group C (8 patients, breast RT size, defined by its greatest dimension, >30 mm); group D (4 patients, RT size ≤30 mm but >20 mm); group E (18 patients, RT size ≤20 mm but not pCR); group F (20 patients, pCR). The only two patients with pCR and a recurrence were integrated into group B (patients 38 and 72, Table 1 and Supplementary Fig. 1). This arbitrary classification was used as we could not determine residual cancer burden in all cases because of a lack of complete breast and lymph node material in some cases treated outside of Centre Jean Perrin (external participants in the trials).
Interpretable targeted panel sequencing (MSK-IMPACT) data were obtained for 45 patients, 26 from the PTMB trial and 19 from the CTX trial. For all patients who reached pCR (n = 13), only the pre-therapy sample was assessed, whereas pre-NAT and post-NAT matched samples were available in 17 of the 32 non-pCR patients. For 15 non-pCR patients, only the post-NAT sample was analyzed (Table 2).
Gene expression analysis by gene arrays was performed in 41 pre-NAT samples, whereas NanoString technology was used to analyze 28 post-NAT samples (Table 2 and Supplementary Fig. 1).
Tumor genomic features
As expected, the vast majority of the tumors carried aberrations in TP53 (44/45) (Fig. 1), which is consistent with the TNBC described genotype24. Interestingly, no mutations were found in one post-NAT sample from group A (patient 49, Table 1). In general, we observed copy number heterogeneity among the groups, with group F being an exception with very low copy number alterations (CNA) (Fig. 2), suggesting that genomic instability does not play a role in determining the aggressiveness of these tumors.

Samples are shown as columns and are ordered by pre/post status and by the group. Genes that were altered in ≥2 samples are shown with colored squares corresponding to the colors in the legend. A rapidly recurring tumors (<1 year post-surgery); (B) tumors with standard recurrences (1–5 years after surgery); (C, D, and E) tumors without pathological complete response (pCR) to neoadjuvant treatment (NAT) and without recurrences; (F) tumors with pCR to NAT and without recurrences. Samples are presented by MSK-IMPACT assay identifiers (X-axis of the heatmap, identifiers of type tnbc-123-A or tnbc-123-AB).

A heatmap shows the amplifications, gains, losses, and homozygous deletions across all samples.
We observed a slight increase in mutations in the post-treatment samples, but this was not statistically significant (Fig. 1). Interestingly, among the 8 most mutated genes after TP53, 5 encode for epigenetic regulators: KMT2D, KDM6A, SETD2, NSD1, and MGA.
Tumor gene expression profiles
In the study of Masuda et al.25, TNBC subtypes are associated with patient outcomes. In our study, we classified patients into 6 subtypes suggested by Lehmann et al.26, based on gene array data (pre-treatment samples), to examine the enrichment of TNBC subtypes in our patient groups. However, we did not observe an enrichment of specific TNBC subtypes in any of our patient groups (Supplementary Fig. 2), suggesting that TNBC subtypes cannot be used to predict patient outcomes in this cohort. Further, there was no statistically significant difference in baseline gene expression between the tumors which recurred (groups A–B) and those which did not (groups C-F). However, differential gene expression analysis, comparing group A tumors with group F tumors (Supplementary Fig. 3), identified 12 HLA genes, which were significantly downregulated in group A. Moreover, all 6 MHC I class and 13 MHC II class HLA genes had lower expression in group A tumors (Fig. 3a), indicating that these tumors would elicit a low immune response. It should be noted that one of group A patients, patient 71 (Fig. 3a, Table 1, respectively), shows high expression in the MHC I class, presenting heterogeneity of HLA expression.

a Heatmap of gene expression in the HLA family. Differentially expressed genes identified from A vs. F are marked in bold. Z-score of expression level was calculated for each gene across all 41 patients. Samples are presented with their original institutional identifiers. A–F, recurrence groups; BL1, basal-like 1; BL2, basal-like 2, IM, immunomodulatory; LAR, luminal androgen receptor; M, mesenchymal; MSL, mesenchymal stem-like; UNS, unclassified. Samples are presented by MSK-IMPACT assay identifiers (X-axis of the heatmap, identifiers of type tnbc-123-A or tnbc-123-AB). b Immune cytolytic activity (CYT). p-value between group A tumors and others was calculated using a two-tailed Student’s t-test.
To confirm the low intratumor immune activity of group A tumors, we calculated the CYT score, a hallmark of cytolytic activity of the tumor immune infiltrate, as proposed by Rooney et al.27. As shown in Fig. 3b, group A tumors show significantly lower CYT scores (P-value <0.03, 95% CI: −3.72, −0.47) in comparison with the tumors with better prognosis, indicating that the intratumor immune activity in the group A tumors is indeed decreased, which might be one of the features permitting their rapid progression after therapy.
As IFN-γ is a key cytokine produced by the immune cells during the anti-tumor immune response, we examined the expression of the IFN-γ-related genes28 and found that, except for patient FAL-SA (patient 71, Table 1), another two patients in group A had extremely low baseline expression of many IFN-γ-related and other immune genes (Fig. 4). Together with low HLA gene expression and CYT scores, these findings reveal a “cold” immunological profile of the rapidly recurring tumors, which existed already before the NAT.

Z-score of expression level was calculated for each gene across all 41 patients. Samples are presented with their original institutional identifiers. A–F, recurrence groups; BL1 basal-like 1, BL2 basal-like 2, IM immunomodulatory, LAR luminal androgen receptor; M mesenchymal, MSL mesenchymal stem-like; UNS unclassified.
Through gene ontology (GO) enrichment analysis in “Molecular Function” terms, we observed 7 out of the top 10 enriched GO terms are related to downregulated cytokine and chemokine activities, including cytokine receptor activity (GO:0004896), cytokine binding (GO:0019955), cytokine activity (GO:0005125), cytokine receptor binding (GO:0005126), chemokine receptor binding (GO:0042379), and chemokine activity (GO:0008009) (Supplementary Fig. 4a). MHC protein complex binding (GO:0023023) is also enriched in downregulated genes, consistent with low HLA gene expression in group A tumors described above. Reactome pathway enrichment analysis identified enriched pathways in downregulated genes, for example, “chemokine receptors bind chemokines”, “interferon-gamma signaling”, “signaling by interleukins”, and so on (Supplementary Fig. 4b). Their pathway networks indicate that these pathways are immune-related (Supplementary Fig. 4c).
Another important difference in the baseline gene expression between group A and group F tumors was significantly higher expression of the cell cycle-related genes in group A. By gene set enrichment analysis we saw that the pathways enriched in those upregulated genes were “Polo-like kinase-mediated events”, “cyclin A/B1/B2 associated events during G2/M transition”, and “Chk1/Chk2(Cds1) mediated inactivation of Cyclin B: Cdk1 complex” (Supplementary Fig. 5). We further performed reactome gene set enrichment analysis using expression fold change values calculated from differential expression analysis between A and F and observed that cell cycle-related pathways are enriched in upregulated genes, including genes involved in different stages of the cell cycle (Fig. 5, left), for example, “S phase” and “separation of sister chromatids”, and genes involved in the regulation of cell cycle (Fig. 5, right), for example, “cell cycle checkpoints” and “mitotic spindle checkpoint”.

On x-axis, genes are ranked based on log2 fold change of gene expression level. From top to bottom, they are enrichment score of pathways on each gene, log2 fold change of expression level.
To examine genes related to metastasis, we compared gene expression data produced by the NanoString gene panel assessing genes involved in cancer progression, in post-NAT residues of tumors with and without recurrences. As shown in Supplementary Fig. 6, we observed that SOX2, a typical embryonic/stemness gene, and CXCR4, a well-known metastasis-related gene, are upregulated in group A tumors; AREG, encoding for amphiregulin, was upregulated in group B tumors. These data suggest that SOX2, CXCR4, and AREG may have a role in TNBC progression after NAT.
Tumor-infiltrating lymphocytes (TILs)
In the RT of patients from group A, TILs were strikingly low before NAT ( ≤5% in all but one patient), which was concordant with gene expression showing low intratumor immune activity. Moreover, no patients in group A had increased TILs after NAT (Table 2). In group B, the baseline TIL density was also low (<1%) in 4 out of 6 evaluated cases. As in group A, no increase in TILs was observed after NAT, but 2 patients from this group reached pCR. Interestingly, one of the pCR cases had 100% TILs before NAT; however, she was diagnosed with a brain metastasis 23 months post-NAT and died a month after (patient 72, Table 2).
Most pCR cases had moderate or high amounts of TILs: 18 out of 22 cases had ≥20% of TILs before NAT, and 10 cases corresponded to the lymphocyte-predominant breast cancer (≥50% TILs) (Table 2). Two out of 23 pCR cases recurred; interestingly, both were very rich in TILs (80–100%) and both developed a brain metastasis, one of which was fatal.
These data indicate that very low TIL density (<5%), before NAT or in the post-NAT RT, could be an indicator of metastatic progression, however if associated with gene expression profile showing low anti-tumor immune activity, as seen in group A.
Discussion
In this work, we present results obtained by molecular analyses of TNBC tissues sampled before and after NAT containing anti-EGFR antibodies and chemotherapy.
In search of molecular features associated with patient outcome, we first observed a marked heterogeneity of the analyzed samples in terms of mutation and CNA profile, without any specific alteration associated with metastatic recurrences. This is in line with other reports where they detect potentially actionable mutations in TNBC tissue but are unable to predict disease recurrence6. These findings may suggest that mutational profiling of TNBC is not the method of choice for the discovery of recurrence biomarkers and that other molecular analyses should be preferentially used for that purpose.
By analysis of gene expression, we distinguished a set of alterations characterizing TNBCs which progressed very rapidly after NAT completion. All but one of the six patients carrying those tumors died of metastatic disease in less than 6 months after surgery (data not shown). Rapid post-NAT progressors are relatively rare among TNBCs (3–5%, personal communication from Dr Mouret-Reynier, Centre Jean Perrin) and typically are undistinguishable from other non-responders in clinical trials. Our study showed that TNBCs with rapid metastatic progression after NAT containing anti-EGFR and chemotherapy had, already before therapy, two important biological characteristics: very low expression of several HLA class I or II genes and high expression of cell cycle-related genes.
MHC/HLA class I loss or downregulation is one of the main mechanisms of cancer immune escape, resulting in decreased T-cell cytolytic anti-tumor response29. In our series, the rapidly recurring tumors had both low expression of MHC I and -II genes and low cytolytic activity scores, indicating the absence of the anti-tumor immune reaction. In breast cancer in general, loss of HLA-I is associated with poor prognosis30,31. This was demonstrated also in TNBC, where higher expression of HLA class 2 molecules is linked to significantly longer disease-free survival16,32. One group has recently developed a 36-gene assay based on Nanostring technology, the MHCII Immune Activation Assay, which identifies TNBC patients with low risk of recurrence; patients with these tumors can be treated by immunotherapy instead of classical cytotoxic therapy16. Similarly, high expression of a gene signature containing HLA-DRA, HLA-E, IDO1, CXCL9, CXCL10, STAT1, and GZMB was found to be associated with TNBCs without relapse in 5 years after NAT33.
Besides low expression of the HLA genes, the rapidly recurring tumors in our series were characterized also by low expression of the IFN-γ signature and several other genes involved in the immune response within the tumor site. Most of those tumors had a very low amount of TILs before NAT and none of them increased TILs after NAT. This could explain their high resistance to the administered treatment, considered as capable of inducing immunogenic cell death and increasing tumor infiltration by cytolytic lymphocytes. Two recent studies proposed that the immune infiltration of post-NAT TNBC residues can influence the metastatic evolution of the disease. Patients with TNBC highly infiltrated by lymphocytes after NAT have longer metastasis-free survival than the ones having post-NAT residues with reduced amounts of TILs34. Moreover, low levels of post-NAT TILs in TNBC were shown to be associated with activation of the RAS/MAPK pathway, one of the proposed mechanisms of resistance to chemotherapy and metastatic progression of TNBC35. Thus, our data indicate that TNBC with very low TILs at the baseline and after NAT may represent a potentially aggressive category.
We also showed that the rapidly recurring tumors had a baseline expression of genes involved in the regulation of the cell cycle much higher than the tumors which experienced pCR to NAT and never recurred, but also higher than any other group in our cohort (data not shown). High pre-NAT proliferation, measured by the Ki67 index, has mostly been reported as a predictor of good response (pCR) to NAT;36 however, very high Ki67 indices (≥50%) were associated shown to be associated with progressive disease37,38. It has been postulated that activation of multiple receptor tyrosine kinases (RTKs) such as EGFR, IGF1R, or MET is responsible, at least in part, for the high proliferation of the progressive TNBCs26. In such cases, blockade of several RTKs might be more efficacious than single-type receptor blockade.
Our findings indicate that simultaneous assessment of the expression of genes involved in tumor proliferation and those involved in the immune response might be an effective way to stratify TNBC patients. In a series of 1954 breast cancers of all molecular subtypes, high expression of a proliferation-related signature and of all three immune metagenes was associated with longer distant metastasis-free survival (DMFS). However, in those highly proliferative breast cancers, low expression of any of the immune metagenes was associated with short DMFS despite high expression of the other two39. These data support the high importance of strong immune control of the highly proliferating breast cancers for a good outcome. On the other hand, the association of high proliferation and immunological silence would favor rapid tumor progression, as observed in our patient cohort.
Our analysis of the post-NAT residues, although very limited, highlighted two pro-metastatic genes, SOX2 and CXCR4, as potential recurrence predictors. SOX2 has been shown to promote proliferation and metastasis in TNBC40. Similarly, several previous studies have shown an association between high CXCR4 expression and breast cancer metastasis41. Interestingly, a recent study showed a strong correlation between high CXCR4 expression and high TIL count42, whereas in our study high CXCR4 expressors were devoid of TILs. More work is therefore necessary to fully explore the relationship between SOX2, CXCR4, and the immune microenvironment of TNBC, however these three parameters seem to have the capacity to predict patient outcomes in TNBC.
Besides potential biomarkers of rapid recurrence after NAT, we did not reveal biomarkers that could stratify the entire cohort into prognostic groups. This likely can be explained by a small number of recurrences as well as of patients/samples analyzed and the heterogeneity of the cohort. These characteristics of the cohort we analyzed here are surely the major limitations of the study. However, since the trials we conducted were the first trials of neoadjuvant EGFR blockade in TNBC, we wanted to analyze the available tissue material, even limited, to generate at least some hypotheses. Another limitation of this study was our inability to calculate residual cancer burden43 because of the lack of the necessary material in some surgical specimens (due to the multicentricity of the clinical trials). It would have been interesting to see whether any of the evaluated molecular features had a specific relationship with any of the RCB categories.
In conclusion, we found that TNBC with high baseline expression of cell cycle genes, associated with a “cold” immune microenvironment, may have a very poor prognosis following neoadjuvant treatment containing chemotherapy and anti-EGFR antibodies. Therefore, high pre-treatment proliferation and low intratumor immune response are worth evaluation in larger studies, on cohorts treated by various neoadjuvant treatments, to verify their general and treatment-specific prognostic value.
Methods
Patients and tumor samples
The available material (formalin-fixed, paraffin-embedded (FFPE) tumor tissues or extracted DNA) from the patients enrolled in clinical trials NCT00933517 (panitumumab trial, PTMB) and NCT00600249 (cetuximab trial, CTX) was used. Details on the inclusion criteria, treatment, response, and non-genomic predictive biomarkers are previously published22,23. The clinical follow-up of 5 years after surgery ended in August 2017. Recurrence was defined as the appearance of any invasive cancer deposit. All local and most distant recurrences were histologically confirmed, except brain metastases which were diagnosed by imaging.
The greatest dimension of post-NAT breast residual tumor and the number of involved axillary lymph nodes were retrieved from the pathology reports received from the participating centers in the trials. The tumors were all but one of Scarff-Bloom-Richardson (SBR) grade 3. The only tumor of SBR grade 2 was an apocrine carcinoma in group E. No SBR grade change was observed after the NAT, in comparison to the pre-NAT grade.
Quantity of tumor-infiltrating lymphocytes (TILs) was estimated on H&E-stained FFPE pre-NAT and post-NAT tumor tissue sections, according to the recommendations of the International Immuno-Oncology Biomarker Working Group on Breast Cancer44,45.
Targeted exome sequencing
DNA samples extracted from formalin-fixed, paraffin-embedded TNBCs (n = 62) were subjected to MSK-IMPACT targeted sequencing at the MSKCC Integrated Genomics Operation as previously described46. Briefly, raw sequence reads were aligned to the reference human genome GRCh37 using the BURROWS-WHEELER ALIGNER (BWA 0.7.15)47. Local realignment, duplicate read removal, and base quality score recalibration were performed using the GENOME ANALYSIS TOOLKIT (GATK 3.7)48. Somatic single nucleotide variants (SNVs) were called using MUTECT (1.1.7)49, and small insertions and deletions (indels) were identified using STRELKA (1.0.15)50, VARSCAN2 (2.3.7)51, LANCET (1.0.0)52, and SCALPEL (0.5.3)53 and further curated by manual inspection. SNVs and indels outside of target regions were filtered out, as were SNVs and indels for which the variant allele fraction (VAF) in the tumor sample was <5 times that of the paired normal VAF as previously described46,54. Finally, SNVs and indels found at >5% global minor allele frequency in dbSNP (build 137) and >5% global allele frequency in EXAC (0.3.1) were discarded. Somatic copy number alterations and loss of heterozygosity were obtained using FACETS55. The cancer cell fractions (CCF) of all mutations were computed using ABSOLUTE (1.0.6)56. A mutation was classified as clonal if its probability of being clonal was >50%57 or if the lower bound of the 95% confidence interval of its CCF was >90%46,54. Mutations that did not meet the above criteria were considered subclonal. A combination of in silico functional predictors was used to define the potential functional impact of each missense SNV as previously described46,58. Mutation hotspots were assigned according to Chang et al.59.
Gene expression by gene arrays
Nucleic acids were extracted from frozen tumor tissue (pre-NAT samples) using AllPrep DNA/RNA mini kit (Qiagen France SAS, Courtaboeuf, France) according to the manufacturer’s protocol. RNA quality was verified using the 2100 BioAnalyzer (Agilent Technologies). The extracted material was sent to Helixio (Saint-Beauzire, France), where it was hybridized with gene arrays (Human SurePrint, Agilent Technologies France, Les Ullis, France). The raw data were transferred to Memorial Sloan Kettering Cancer Center for bioinformatics and statistical analysis.
Limma package60 was applied to process Agilent microarray data and identify differentially expressed genes between progressive tumors and tumors with pathological complete response. Background correction was performed on expression intensities before normalization by the parameter of “expnorm”. The median value was taken when a gene has multiple probes. Then, “cyclicloess” method, which applies loess normalization to all possible pairs of arrays, was used to normalize corrected intensities. Fold change of expression intensities and P-values were estimated, which were used to identify differentially expressed genes.
We performed GO61 and Reactome pathway62 enrichment analysis with clusterProfiler63 and ReactomePA64. Over-representation analysis and gene set enrichment analysis were performed. Benjamini-Hochberg method was used to calculate adjusted p-values.
TNBCtype was used to classify samples into transcriptomic subtypes26,65.
Immune cytolytic activity of the local immune infiltrate was quantified by measuring expression of the granzyme A (GZMA) and perforin (PRF1) genes, as proposed by Rooney et al.27. We calculated the CYT score by averaging the expression level of GZMA and PRF1 for each microarray sample66.
Gene expression by NanoString
RNA was isolated from FFPE samples of post-NAT residual tumors using the High Pure FFPET RNA Isolation Kit (Roche Diagnostics, Meylan, France), according to the manufacturer’s instructions. RNA purity and concentration were determined using a spectrophotometer (BioPhotometer, Eppendorf). Only the samples with a purity of ≥1.6 were used. One hundred nanograms of RNA was loaded for hybridization with nCounter® PanCancer Progression Panel, according to the manufacturer’s instructions (NanoString, Seattle, WA, USA). The hybridization signals were analyzed by the nCounter® FLEX Analysis System (NanoString).
Raw count data, which include 740 endogenous genes, 6 positive controls, 8 negative controls, and 30 housekeeping genes, were preprocessed using the R package NanoStringNorm67. Raw counts were merged by patients by taking an average of samples. Specifically, geometric mean-based normalization was used to normalize for technical assay variation, followed by background adjustment based on negative controls, where “mean.2sd” is used. RNA content normalization was performed by all 30 housekeeping genes with the parameter “housekeeping.geo.mean”. Finally, log2-transformed data were used for downstream analysis. One sample was excluded for the analysis due to the high positive normalization factor examined by NanoStringNorm67. Differential expression analysis was performed using the R package Limma60.
Statistical analysis
The statistical significance of CYT differences between group A and other tumors was evaluated by a two-tailed Student t test in the R package.
Ethical approval
This study was declared to and approved by the French National Commission for Informatics and Freedom (Commission Nationale de l’Informatique et des Libertés), under number 1209138, as well as by the Ethical Committee of Clermont Ferrand (numbers AU 806 and 711). Each patient signed an informed consent presenting details of the study.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The assembled prospective somatic mutational data from ctDNA and tumors for the entire cohort have been deposited for visualization and download in the cBioPortal for Cancer Genomics (http://cbioportal.org/). MSK-IMPACT DNA sequencing data is available under SRA accession code PRJNA750756, gene array RNA sequencing data is available under GEO accession code GSE180775 and nanostring sequencing data is available under GEO accession code GSE180717. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
References
Bianchini, G., Balko, J. M., Mayer, I. A., Sanders, M. E. & Gianni, L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690 (2016).
Geyer, F. C. et al. The spectrum of triple-negative breast disease: high- and low-grade lesions. Am. J. Pathol. 187, 2139–2151 (2017).
Kennecke, H. et al. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 28, 3271–3277 (2010).
Dent, R. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 13, 4429–4434 (2007).
Cortazar, P. et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164–172 (2014).
Balko, J. M. et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Disco. 4, 232–245 (2014).
Denkert, C. et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet. Oncol. 19, 40–50 (2018).
Luen, S. J. et al. Prognostic implications of residual disease tumor-infiltrating lymphocytes and residual cancer burden in triple negative breast cancer patients after neo-adjuvant chemotherapy. Ann. Oncol. https://doi.org/10.1093/annonc/mdy547 (2018).
Burstein, M. D. et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 21, 1688–1698 (2015).
Lehmann, B. D. et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One 11, e0157368 (2016).
Yu, K. D. et al. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin. Cancer Res. 19, 2723–2733 (2013).
Yau, C. et al. An optimized five-gene multi-platform predictor of hormone receptor negative and triple negative breast cancer metastatic risk. Breast Cancer Res. 15, R103 (2013).
Lee, U. et al. A prognostic gene signature for metastasis-free survival of triple negative breast cancer patients. PLoS One 8, e82125 (2013).
Qian, J. et al. A 3q gene signature associated with triple negative breast cancer organ specific metastasis and response to neoadjuvant chemotherapy. Sci. Rep. 7, 45828 (2017).
Pece, S. et al. Identification and clinical validation of a multigene assay that interrogates the biology of cancer stem cells and predicts metastasis in breast cancer: a retrospective consecutive study. EBioMedicine 42, 352–362 (2019).
Stewart, R. L. et al. A multigene assay determines risk of recurrence in patients with triple-negative breast cancer. Cancer Res. 79, 3466–3478 (2019).
Axelrod, M. L. et al. Changes in peripheral and local tumor immunity after neoadjuvant chemotherapy reshape clinical outcomes in patients with breast cancer. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-19-3685 (2020).
Economopoulou, P., Kaklamani, V. G. & Siziopikou, K. The role of cancer stem cells in breast cancer initiation and progression: potential cancer stem cell-directed therapies. Oncologist 17, 1394–1401 (2012).
Chang, Y. S., Jalgaonkar, S. P., Middleton, J. D. & Hai, T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc. Natl Acad. Sci. USA 114, E7159–E7168 (2017).
Samanta, D. et al. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc. Natl Acad. Sci. USA 115, E1239–E1248 (2018).
Karagiannis, G. S. et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aan0026 (2017).
Nabholtz, J. M. et al. Multicentric neoadjuvant phase II study of panitumumab combined with an anthracycline/taxane-based chemotherapy in operable triple-negative breast cancer: identification of biologically defined signatures predicting treatment impact. Ann. Oncol. 25, 1570–1577 (2014).
Nabholtz, J. M. et al. Multicentric neoadjuvant pilot Phase II study of cetuximab combined with docetaxel in operable triple negative breast cancer. Int. J. Cancer 138, 2274–2280 (2016).
Shah, S. P. et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486, 395–399 (2012).
Masuda, H. et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 19, 5533–5540 (2013).
Lehmann, B. D. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767 (2011).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015).
Danaher, P. et al. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 6, 63 (2018).
Aptsiauri, N., Ruiz-Cabello, F. & Garrido, F. The transition from HLA-I positive to HLA-I negative primary tumors: the road to escape from T-cell responses. Curr. Opin. Immunol. 51, 123–132 (2018).
Kaneko, K. et al. Clinical implication of HLA class I expression in breast cancer. BMC Cancer 11, 454 (2011).
Park, H. S. et al. Loss of human leukocyte antigen class I expression is associated with poor prognosis in patients with advanced breast cancer. J. Pathol. Transl. Med. 53, 75–85 (2019).
Pedersen, M. H. et al. Downregulation of antigen presentation-associated pathway proteins is linked to poor outcome in triple-negative breast cancer patient tumors. Oncoimmunology 6, e1305531 (2017).
Perez-Pena, J. et al. A transcriptomic immunologic signature predicts favorable outcome in neoadjuvant chemotherapy treated triple negative breast tumors. Front. Immunol. 10, 2802 (2019).
Dieci, M. V. et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: a retrospective multicenter study. Ann. Oncol. 25, 611–618 (2014).
Loi, S. et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 22, 1499–1509 (2016).
Chen, X. et al. The predictive value of Ki-67 before neoadjuvant chemotherapy for breast cancer: a systematic review and meta-analysis. Future Oncol. 13, 843–857 (2017).
Caudle, A. S. et al. Predictors of tumor progression during neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 28, 1821–1828 (2010).
Tanabe, Y. et al. Pathological features of triple-negative breast cancers that showed progressive disease during neoadjuvant chemotherapy. Cancer Sci. 108, 1520–1529 (2017).
Nagalla, S. et al. Interactions between immunity, proliferation and molecular subtype in breast cancer prognosis. Genome Biol. 14, R34 (2013).
Liu, P. et al. SOX2 promotes cell proliferation and metastasis in triple negative breast cancer. Front. Pharm. 9, 942 (2018).
Zhang, Z. et al. Expression of CXCR4 and breast cancer prognosis: a systematic review and meta-analysis. BMC Cancer 14, 49 (2014).
Lyu, L. et al. Comprehensive analysis of the prognostic value and immune function of chemokine-CXC receptor family members in breast cancer. Int. Immunopharmacol. 87, 106797 (2020).
Symmans, W. F. et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J. Clin. Oncol. 25, 4414–4422 (2007).
Salgado, R. et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann. Oncol. 26, 259–271 (2015).
Dieci, M. V. et al. Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: a report of the International Immuno-Oncology Biomarker Working Group on Breast Cancer. Semin Cancer Biol. 52, 16–25 (2018).
Ng, C. K. et al. Intra-tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol. 16, 107 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Saunders, C. T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Narzisi, G. et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun. Biol. 1, 20 (2018).
Narzisi, G. et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat. Methods 11, 1033–1036 (2014).
Weigelt, B. et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J. Natl Cancer Inst. 110, 1030–1034 (2018).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 44, e131 (2016).
Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
Landau, D. A. et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726 (2013).
Martelotto, L. G. et al. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 15, 484 (2014).
Chang, M. T. et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 (2016).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Gene Ontology, C. Gene ontology consortium: going forward. Nucleic Acids Res. 43, D1049–1056 (2015).
Fabregat, A. et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 44, D481–487 (2016).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Yu, G. & He, Q. Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 12, 477–479 (2016).
Chen, X. et al. TNBCtype: a subtyping tool for triple-negative breast cancer. Cancer Inf. 11, 147–156 (2012).
Tian, T. et al. A comprehensive survey of immune cytolytic activity-associated gene co-expression networks across 17 tumor and normal tissue types. Cancers https://doi.org/10.3390/cancers10090307 (2018).
Waggott, D. et al. NanoStringNorm: an extensible R package for the pre-processing of NanoString mRNA and miRNA data. Bioinformatics 28, 1546–1548 (2012).
Acknowledgements
This work was founded by the NIH grant P30CA008748, the Breast Cancer Research Foundation, the Geoffrey Beene Cancer Research Center.
Author information
Authors and Affiliations
Contributions
N.R.-R., J.S.R.-F. and M.S. designed the experiment. N.R.-R., P.S. and Y.Z. performed the analyses and generated the data. N.R.-R., P.S., Y.Z., H.H.W., M.H.B., L.F., E.C., F.P.-C. analyzed the data. M.P., C.A., J.-M.N. and F.P.-L. provided clinical support of the study. N.R.-R., P.S. and Y.Z. drafted the paper. All authors reviewed and approved the final form of the paper and all attachments.
Corresponding authors
Ethics declarations
Competing interests
M.S. has received research funds from Puma Biotechnology, AstraZeneca, Daiichi-Sankyo, Immunomedics, Targimmune, and Menarini Ricerche, is a cofounder of Medendi.org and is on the advisory board of Menarini Ricerche. M.S. is an employee and stockholder of AstraZeneca. J.S.R.-F. is a consultant of Goldman Sachs and REPARE Therapeutics, a member of the Scientific Advisory Board of VolitionRx and Paige.AI, and an ad hoc member of the Scientific Advisory Board of Ventana Medical Systems, Roche, Genentech, Novartis, and InviCRO, outside of the scope of the submitted work. F.P.-L. has received consulting honoraria from Merck, Amgen, and Sanofi. All the other authors do not report potential conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Radosevic-Robin, N., Selenica, P., Zhu, Y. et al. Recurrence biomarkers of triple negative breast cancer treated with neoadjuvant chemotherapy and anti-EGFR antibodies. npj Breast Cancer 7, 124 (2021). https://doi.org/10.1038/s41523-021-00334-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-021-00334-5
