
Abstract
The development of acquired drug resistance hampers the long-term success of B-RAF inhibitor therapy for melanoma patients. Here we show V600EB-RAF copy-number gain as a mechanism of acquired B-RAF inhibitor resistance in 4 out of 20 (20%) patients treated with B-RAF inhibitor. In cell lines, V600EB-RAF overexpression and knockdown conferred B-RAF inhibitor resistance and sensitivity, respectively. In V600EB-RAF amplification-driven (versus mutant N-RAS-driven) B-RAF inhibitor resistance, extracellular signal-regulated kinase reactivation is saturable, with higher doses of vemurafenib down-regulating phosho-extracellular signal-regulated kinase and re-sensitizing melanoma cells to B-RAF inhibitor. These two mechanisms of extracellular signal-regulated kinase reactivation are sensitive to the MEK1/2 inhibitor AZD6244/selumetinib or its combination with the B-RAF inhibitor vemurafenib. In contrast to mutant N-RAS-mediated V600EB-RAF bypass, which is sensitive to C-RAF knockdown, V600EB-RAF amplification-mediated resistance functions largely independently of C-RAF. Thus, alternative clinical strategies may potentially overcome distinct modes of extracellular signal-regulated kinase reactivation underlying acquired B-RAF inhibitor resistance in melanoma.
Similar content being viewed by others

Introduction
Activating B-RAF V600 kinase mutations occur in ~50% of melanomas1, and the ATP-competitive type I RAF inhibitors, PLX4032/vemurafenib and GSK2118436, display remarkable anti-tumour activity leading to overall survival advantage in patients with V600B-RAF mutant melanomas2,3,4,5,6. Acquisition of drug resistance leading to clinical relapse, however, develops in virtually all patients treated with B-RAF inhibitors (B-RAFi)4,5. Heterogeneous mechanisms of acquired B-RAFi resistance hitherto uncovered fall into general mitogen-activated protein kinase (MAPK)-redundant, AKT-dependent7,8 or MAPK-reactivating9,10 pathways, indicating specific translatable therapeutic strategies to prevent or overcome resistance. Contrary to expectation, V600EB-RAF secondary mutations have not been found to account for acquired B-RAFi resistance10, suggesting V600EB-RAF-bypass mechanisms as the principal means to extracellular signal-regulated kinase (ERK) reactivation.
Here we observed an alteration in V600EB-RAF, namely genomic copy-number gain, in tumours of melanoma patients whose cancer progressed after initial responses to B-RAFis. We demonstrated that this V600EB-RAF amplification results in V600EB-RAF overexpression, which is necessary and sufficient for acquired resistance to B-RAFi. This finding, along with a recent study reporting amino-terminal truncation of V600EB-RAF causing acquired B-RAFi resistance in melanoma11, underscores key molecular alterations in the drug target itself. We further suggest that V600EB-RAF-instrinsic (overexpression, truncation) versus V600EB-RAF-bypass (N-RAS mutations) mechanisms, both reactivating the MAPK pathway, may offer insights into distinct therapeutic strategies to overcome acquired B-RAFi resistance in melanoma.
Results
Whole-exome sequencing identifies V600EB-RAF amplification.
We assembled 20 sets of patient-matched baseline (before B-RAFi therapy) and disease progression (DP; that is, acquired B-RAFi resistance) melanoma tissues and analysed them to identify the proposed mechanisms of acquired B-RAFi resistance in melanoma. These reported mechanisms include N-RAS10 and MEK1 (ref. 12) mutations, alternative-spliced V600EB-RAF variants11, and overexpression of receptor tyrosine kinases (RTKs) (PDGFRβ7,10, IGF1-R8) and COT9 (Tables 1 and Supplementary Table S1 and Fig. S1). For DP samples negative for these mechanisms and where there was sufficient frozen and patient-matched normal tissues (from patients no. 4, 5, 8, 14, 16, 17 and 18), we subjected triads of genomic DNAs (gDNAs) from normal, baseline and DP tissues to whole-exome sequencing. In two available data sets, we searched for somatic DP-specific non-synonymous single-nucleotide variants (snSNVs) and small insertion-deletion (indels), which were exceedingly few in number or absent, respectively, using our bioinformatic workflow (Supplementary Tables S2 and S3). We also analysed for DP-specific copy-number variations (CNVs) from the exome sequence data (Supplementary Table S2). This identified V600EB-RAF copy-number gains in these two patients' DP tissues (2.2- and 12.8-fold in patients no. 5 and 8, respectively) relative to their respective baseline tissues (Fig. 1a; Table 1). Gain in V600EB-RAF copy number was reflected in corresponding increased gene expression at the protein level (Fig. 1b).

(a) Copy-number variations (CNVs) called from whole-exome sequence data on two triads of gDNAs using ExomeCNV and chromosome 7 as visualized by Circos (outer ring, genomic coordinates (Mbp); centromere, red; inner ring, log ratio values between baseline and disease progression (DP) samples' average read depth per each capture interval; scale of axis for patient no. 5 –5 to 5 and for patient no. 8 –2.5 to 2.5). Two patients whose melanoma responded to and then progressed on vemurafenib. The genomic region coded orange (magnified views shown in the center of Circos maps) represents the location of B-RAF (chr7:140,424,943-140,524,564), which shows an average log ratio value of 1.14 (2.2-fold gain; patient no. 5) and 3.8 (12.8-fold gain; patient no. 8). (b) B-RAF immunohistochemistry on paired tissues derived from the corresponding patients as in a (scale bar, 50 μM). (c) Validation of V600EB-RAF copy-number gain by gDNA Q–PCR (black and red by B-RAF primer set 1 and 2, respectively) and recurrence across distinct patients (positives highlighted in orange). PMN, peripheral mononuclear cells; HDF, human dermal fibroblasts for diploid gDNAs. (d) B-RAF V600 mutant to WT ratio increases with disease progression or acquisition of B-RAFi resistance mediated by mutant B-RAF copy-number gain. Chromatograms from Sanger sequencing for melanoma samples from patients who acquired B-RAFi resistance based on distinct molecular alterations: V600EB-RAF copy-number gain, V600EB-RAF truncation, N-RAS mutation or RTK overexpression.
V600EB-RAF amplification was validated by gDNA quantitative PCR (Q–PCR), producing consistent fold increases in DP-specific V600EB-RAF copy-number gain (relative to baseline; 2.0- and 14-fold increase in patient no. 5 and 8, respectively; Fig. 1c). We then expanded the analysis of V600EB-RAF amplification to all 20 paired melanoma tissues and detected V600EB-RAF copy-number gains in DP samples from two additional patients (2.3- and 3-fold for DP2 of patient no. 9 and DP of patient no. 13, respectively; Fig. 1c; Table 1). We note that these copy-number fold increases are likely underestimates of the true changes due to non-tumour diploid cell contents and tumour heterogeneity, as most disease progressive tumours occur from stable residual tumours as a result of partial responses seen in the vast majority of patients treated with B-RAFis. An increase in the mutant B-RAF to wild-type (WT) B-RAF ratio was also noted in all four cases of DP harbouring B-RAF copy-number gain when compared with their respective baseline tissues (Fig. 1d), consistent with selection for V600EB-RAF (versus the WT B-RAF allele) copy-number gain during acquisition of B-RAFi resistance. V600EB-RAF amplification was largely mutually exclusive with N-RAS mutations (no enrichment in MEK1 exon 3 mutation was detected in DP versus baseline tumours), RTK overexpression (no COT overexpression detected), as well as a novel mechanism involving V600EB-RAF alternative splicing11 (Table 1; Supplementary Fig. S1).
B-RAFi selects for V600EB-RAF gain and overexpression.
We have derived vemurafenib/PLX4032-resistant (R) sublines by providing continuous vemurafenib exposure to seven human melanoma-derived V600EB-RAF-positive parental (P) cell lines sensitive to vemurafenib-mediated growth inhibition. Four resistant sublines, including M229 R5 and M238 R1 (refs 7,10), overexpressed PDGFRβ compared with their parental counterpart. One subline (M249 R4 (ref. 10)) gained a mutation in N-RAS, and another (M397 R) an alternatively spliced variant of V600EB-RAF resulting in in-frame fusion of exons 1 and 11 (Supplementary Fig. S2). As in our tissue analysis, these mechanisms were identified in a mutually exclusive manner. Another vemurafenib-resistant subline, M395 R, was derived from a V600EB-RAF-homozygous parental line, M395 P (Supplementary Fig. S3a). Compared with M395 P, M395 R harbours increased copy numbers of V600EB-RAF gDNA and complementary DNA, consistent with a marked V600EB-RAF protein overexpression (Supplementary Fig. S3b,c,d). M395 R displays growth highly resistant to vemurafenib treatment (Supplementary Fig. S4a), and titration of M395 R with vemurafenib (1 h) after a 24 h of drug withdrawal revealed phospho-ERK levels to be highly resistant to acute V600EB-RAF inhibition (Supplementary Fig. S4b). This pattern of MAPK reactivation was similar to that seen in a mutant N-RAS-driven, vemurafenib-resistant subline, M249 R4, and contrasted with that in the RTK-driven vemurafenib-resistant subline, M229 R5 (Supplementary Fig. S4b)7,10. Expectedly, the levels of p-AKT are unchanged (Fig. 2b) comparing M395 P versus M395 R, consistent with a lack of RTK overexpression leading to MAPK-redundant, PI3K-AKT signalling7. Accordingly, M395 R does not overexpress either PDGFRβ or IGF-1R, in contrast to M229 R5, which has been shown to overexpress the RTK PDGFRβ (Supplementary Fig. S4c)7,8. In addition, M395 R is WT for N-, H- and K-RAS and MEK1, harbours no secondary mutations in V600EB-RAF or an alternatively spliced variant of V600EB-RAF, which results in a N-terminally truncated V600EB-RAF protein.

(a,b) Western blot showing that V600EB-RAF overexpression at levels shown did not alter the phospho-ERK (pERK) level in the absence of vemurafenib/PLX4032 (tubulin as loading control) but conferred growth resistance to the parental line, M395 P, when exposed to indicated concentrations of PLX4032 for 72 h (relative to DMSO-treated controls; mean±s.e.m., n=5). Dashed line, 50% inhibition. (c,d) Transduction of shRNA to knockdown BRAFV600E in the drug-resistant subline, M395 R, did not alter the pERK level in the absence of PLX4032 but restored growth sensitivity to PLX4032 (72 h; mean±s.e.m., n=5). (e) Increasing (in M395 P) or decreasing (in M395 R) BRAFV600E levels decreased or increased pERK sensitivity to PLX4032 (0, 0.1, 1, 10 μM) treatments for 1 h, respectively.
V600EB-RAF overexpression correlates with B-RAFi resistance.
Three different but uniformly modest levels of V600EB-RAF overexpression were achieved by infecting M395 P with varying viral titres and subsequent puromycin selection. This resulted in relatively low (1.9-fold over empty vector virus control), medium (2.4-fold) and high (2.8-fold) levels of V600EB-RAF RNA/cDNA overexpression (Supplementary Fig. S5), with the corresponding protein overexpression levels shown in Fig. 2a. In comparison, in two sets of tissues (from patients no. 8 and 13), where flash-frozen tissues were available, the RNA/cDNA levels of V600EB-RAF in the DP tumours were 9.5- and 1.4-fold relative to those in their patient-matched baseline tumours. Notably, the DP tumour from patient no.13 was obtained by an intervention radiology-guided needle biopsy of a pelvic mass (Supplementary Table S1) and contained a high admixture of normal and tumour contents (latter indicated by S100), which likely contributed to an underestimation of the true change in the V600EB-RAF RNA/cDNA levels.
V600EB-RAF overexpression leads to drug-saturable resistance.
The modest and incremental overexpression of V600EB-RAF at the RNA and protein levels in M395 P conferred similar degrees of vemurafenib resistance (Fig. 2b). Interestingly, further V600EB-RAF overexpression at a much greater level, as in the case of M395 R relative to M395 P (increase in RNA/cDNA level shown in Supplementary Figs S3c and S5; increase in protein level shown in Fig. 2c) conferred enhanced drug resistance mainly at 1 μM vemurafenib but not 10 μM vemurafenib (Fig. 2d). Thus, a modest V600EB-RAF copy-number gain and overexpression can confer vemurafenib resistance, and even high amplitude V600EB-RAF amplification and overexpression can be readily saturable by micromolar concentrations of vemurafenib.
Moreover, V600EB-RAF knockdown in M395 R confers vemurafenib sensitivity (Fig. 2c and d). Consistently, V600EB-RAF overexpression in M395 P (at a level titrated to be comparable to M395 R) and its knockdown in M395 R resulted in pERK resistance and sensitivity, respectively, to acute vemurafenib treatment after a 24 h drug withdrawal (Fig. 2e). We predicted that, regardless of the cellular genetic context, MAPK reactivation due to drug target (that is,V600EB-RAF) overexpression would be saturable by higher doses of vemurafenib, in contrast to mutant N-RAS-mediated MAPK reactivation where V600EB-RAF may be bypassed by the alternative use of C-RAF13. Indeed, dosing of vemurafenib from 1 to 50 μM revealed a significant difference in drug sensitivity of M249 R4 (Q61KN-RAS) versus M395 R (amplifiedV600EB-RAF; Fig. 3a; where the latter was highly sensitive to vemurafenib at this drug concentration range), suggesting a potential therapeutic opportunity. To rule out that these results were not due to a difference in genetic backgrounds, we artificially rendered the V600EB-RAF melanoma cell line, M229, vemurafenib-resistant by either Q61KN-RAS or V600EB-RAF viral transduction (Fig. 3b). Again, high dose vemurafenib treatment was more effective at overcoming drug resistance inV600EB-RAF-transduced M229 than in the same cell line transduced with Q61KN-RAS.

(a) Survival curves of B-RAFi-acquired resistant sublines, with indicated mechanisms of resistance, to 72 h of B-RAFi (PLX4032) treatments, showcasing differential responses at the micromolar drug range. Results are shown relative to DMSO-treated controls (mean±s.e.m., n=5; dashed line, 50% inhibition). (b) Survival curves of cell lines, engineered by viral transduction of M229 P to be B-RAFi resistant, to 72 h of B-RAFi (PLX4032) treatments, showcasing differential responses at the micromolar drug range. Results are shown relative to DMSO-treated controls (mean±s.e.m., n=5). Expression of indicated viral expression constructs shown in western blots. (c) Survival curves of B-RAFi-acquired resistant sublines, with indicated mechanisms of resistance, to 72 h of MEKi (AZD6244) treatments, showcasing differential responses at the micromolar drug range. Results are shown relative to DMSO-treated controls (mean±s.e.m., n=5). (d) Survival curves of cell lines (engineered by viral transduction of M229 P and M238 P to overexpress V600EB-RAF rendering these parental cells resistant to B-RAFi) to 72 h of MEKi (AZD6244) treatments, showcasing differential responses at the micromolar drug range. Pt55 R (double B-RAF and N-RAS mutant) is a short-term melanoma culture derived from a tumour, which acquired PLX4032 (vemurafenib) resistance in a treated patient. Results are shown relative to DMSO-treated controls (mean±s.e.m., n=5). (e,f) Indicated cell lines were treated with constant ratios of PLX4032 and AZD6244 and survival measured after 72 h. Relative synergies, expressed as log10 of CI values, are shown. (g) M249 R4 and M395 R were seeded at single-cell density and treated with indicated concentrations of PLX4032 and/or AZD6244. Inhibitors and media were replenished every 2 days, colonies visualized by crystal violet staining after 8 days of drug treatments, and quantified (% growth relative to cells treated with 1 μM PLX4032). Photographs representative of two independent experiments. (h) Survival curves of indicated cell lines after shScrambled or shC-RAF transduction (inset) and when treated with PLX4032 for 72 h. (i) Clonogenic assays of cell lines in e with 14 days (M249 R4) or 18 days (M395 R) of PLX4032 treatment. Results are representative of two experiments.
MEK inhibition restores vemurafenib sensitivity.
As both N-RAS mutation and V600EB-RAF amplification-driven acquired resistance mechanisms would be anticipated to result in MEK reactivation, we tested the allosteric MEK inhibitor (MEKi), AZD6244/selumetinib, on the Q61KN-RAS-driven M249 R4 and the V600EB-RAF amplification-driven M395 R sublines. MEKi treatment resulted in decreased proliferation in both cases, but the activity was noted at lower concentrations for the Q61KN-RAS-driven resistance mechanism (Fig. 3c). This differential pattern was reproducible by exposing AZD6244/selumetinib to V600EB-RAF melanoma cell lines M229 and M238 transduced with high levels of V600EB-RAF versus a short-term culture, Pt55 R10, with Q61KN-RAS-driven acquired B-RAFi resistance (Fig. 3d). We also tested the combination of B-RAFi with MEKi, which is currently in clinical testing14, in 3-day survival assays. A calculation of combination index (CI) values using equal ratios of vemurafenib and selumetinib was performed. The results were consistent with a highly synergistic effect of these two agents combined in overcoming both mutant N-RAS-driven (M249 R4) and V600EB-RAF amplification-driven B-RAFi resistance (M395 R; Fig 3e,f), although the combination tended to be more potent against mutant N-RAS-driven acquired resistance to vemurafenib. This B-RAFi and MEKi combinatorial synergy was further corroborated in longer-term clonogenic assays (Fig. 3g).
Differential C-RAF dependency of ERK-reactivating mechanisms.
We also predicted that MAPK reactivation due to V600EB-RAF overexpression would be C-RAF-independent, in contrast to mutant N-RAS-mediated MAPK reactivation where V600EB-RAF may be bypassed by the alternative use of C-RAF. Indeed, C-RAF knockdown by short hairpin RNA (shRNA) sensitized the mutant N-RAS subline, M249 R4, but not the V600EB-RAF amplified subline, M395 R, to vemurafenib in 3-day survival assays (Fig. 3h). C-RAF knockdown restored vemurafenib sensitivity to M249 R4 (Q61KN-RAS/V600EB-RAF) even more strikingly in a longer-term clonogenic assays, which afforded fresh drug replacement every 2 days (Fig. 3i). An independent C-RAF shRNA also restored vemurafenib sensitivity to M249 R4 (Supplementary Table S4). In addition, B-RAFi and MEKi synergy and C-RAF-dependence in mutant N-RAS-driven acquired B-RAFi resistance was confirmed in a short-term culture derived from a tumour with clinical acquired vemurafenib resistance (Supplementary Fig. S6).
Discussion
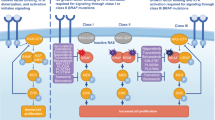
Identification of V600EB-RAF amplification as a mechanism of acquired resistance in B-RAFi-treated patients provides evidence for alterations in the drug target causing clinical relapse. Based on these studies, therapeutic stratification of MAPK reactivation underlying B-RAFi resistance into drug-saturable or C-RAF-dependent pathways may be translatable into the design of next-generation clinical trials aimed at preventing or overcoming B-RAFi resistance (Fig. 4). These findings also provide pre-clinical rationale for dose escalation studies in selected patients with B-RAFi-resistant V600E/KB-RAF metastatic melanomas, particularly given the wide range of effective dosing and the fact that the maximum tolerated dose of GSK2118436 has not been determined. The combination of current B-RAFis (or next-generation RAF inhibitors that enhance B-RAF potency or feature pan-RAF inhibition) with MEK1/2 inhibitors may potentially broadly block MAPK reactivation.

Distinct strategies to overcome acquired resistance driven by amplification of mutant B-RAF or mutations in N-RAS. Schematic of ERK-reactivating pathways (V600EB-RAF amplification indicated by stacked symbols, top; N-RAS mutation, bottom; mutant proteins in red and WT proteins in grey) and proposed strategies to restore B-RAFi sensitivity (increasing B-RAFi concentration or potency, top; switching B-RAFi to pan-RAFi, bottom). Alternatively, the combination of B-RAFi and MEKi are predicted to synergistically growth-inhibit melanomas with acquired resistance to B-RAFi monotherapy stemming from ERK reactivation.
Emerging evidence points to B-RAF mutant cancers of other tissue origin or lineage being less responsive to specific B-RAF inhibition than B-RAF mutant melanomas. Mechanisms of acquired B-RAFi resistance may turn out to be instructive for understanding primary resistance of B-RAF mutant cancer types to B-RAFis, as primary (de novo) and secondary (or acquired) drug resistance may be clinical manifestations from a spectrum of molecular alterations that are mechanistically linked. Thus, multiple modes (for example, mutation, copy-number gain) of up-regulating oncogene activity, which may pre-exist in the same tumour and/or patient, may help to explain the range of heterogeneous responses of B-RAF mutant cancers to direct B-RAF, MEK or ERK inhibition.
Methods
Cell culture experiments
Cells were maintained in DMEM with 10 or 20% fetal bovine serum and glutamine. shRNAs (Supplementary Table S4) for B-RAF and C-RAF were subcloned into the lentiviral vector pLL3.7; pBabe B-RAF (V600E) was purchased from (plasmid 17544) Addgene; viral supernatants generated by co-transfection with three packaging plasmids into HEK293T cells; and infections carried out with protamine sulphate. Stocks and dilutions of PLX4032 (Plexxikon, Berkeley, CA, USA) and AZD6244 (commercially available) were made in DMSO. Cells were quantified using CellTiter-GLO Luminescence (Promega) or crystal violet staining followed by NIH Image J quantification.
Whole-exome sequencing
Human tissues were obtained with patient-informed consent under UCLA Institutional Review Board (no. 10-001089) approval. For each sample, 3 μg of high-molecular-weight gDNA was used as the starting material to generate the sequencing library. Exome captures were performed using Agilent SureSelect Human All Exon 50 mb and Agilent SureSelect Human All Exon 50 mb XT for patient no. 5 and 8, respectively, per manufacturers' recommendation, to create a mean 200 bp insert library. For patient no. 5, sequencing was performed on Illumina GenomeAnalyzerII (GAII) as 76+76 bp paired-end run. The normal sample was run on one flowcell lane and the tumour samples were run on two flowcell lanes each. For patient no. 8, sequencing was performed on Illumina HiSeq2000 as 50+50 bp paired-end run and 100+100 bp paired-end run. The three samples (normal, baseline and DP) were initially mixed with nine other samples and run across five flowcell lanes for the 50+50 bp run. For the 100+100 bp run, they were mixed with three other samples to be run across five flowcell lanes with barcoding of each individual genomic sample library.
For patient no. 5, approximately 62 million, 137 million and 147 million reads were generated for normal tissue (skin), baseline melanoma and DP melanoma, respectively, with 75.2%, 78.1% and 74.7% of the reads mapping to capture targets. Based on an analysis of reads that uniquely aligned to the reference genome and for which the potential PCR duplicates were removed, an average coverage of 52X, 88X and 114X was achieved with 87%, 92% and 93% of the targeted bases being covered at 10X or greater read depth for normal, baseline and DP, respectively.
For patient no. 8, approximately 198 million, 270 million and 256 million reads were generated for normal tissue (skin), baseline melanoma and DP melanoma, respectively, with 43.2%, 44.1% and 42.3% of the reads mapping to capture targets. Based on an analysis of reads that uniquely aligned to the reference genome and for which the potential PCR duplicates were removed, an average read depth of 107X, 132X and 123X was achieved with 89%, 90% and 90% of the targeted bases being covered at 10X or greater for normal, baseline and DP, respectively.
Sequencing data analysis.
For patient no. 8 where the samples were indexed and pooled before the sequencing, Novobarcode from Novocraft was used to de-multiplex the data. The sequence reads were aligned to the human reference genome using Novoalign V2.07.13 from Novocraft (http://www.novocraft.com). For patient no. 5, hg18 downloaded from UCSC genome database was used and for patient no.8, b37 downloaded from GATK (Genome analysis toolkit; http://www.broadinstitute.org/gsa/wiki/index.php/GATK_resource_bundle#b37_resources:_the_standard_data_set) resources website was used for the reference genome. SAMtools v.0.1.16 (ref. 16) was used to sort and merge the data and Picard (http://picard.sourceforge.net/) was used to mark PCR duplicates. To correct the misalignments due to the presence of indels, local realignment was performed using RealignerTargetCreator and IndelRealigner of GATK17. Indel calls in dbSNP132 were used as known indel input. Then, GATK CountCovariates and TableRecalibration were used to recalibrate the originally reported quality score by using the position of the nucleotide within the read and the preceding and current nucleotide information. Finally, to call the single-nucleotide variants (SNVs), the GATK UnifiedGenotyper was used to the realigned and re-calibrated bam file while GATK IndelGenotyperV2 was used to call small insertion/deletions (Indels). To generate a list of somatic variants for DP tumour, the difference in allele distribution was calculated using one-sided Fisher's exact test using normal sample or the baseline sample. Variants with P-value <0.05 were included in the 'somatic variant list'. Low coverage (<10X) SNVs and SNVs with more than one variant allele in normal tissue and baseline melanoma were filtered out during the process. These somatic variants were further annotated with SeattleSeqSNPannotation (http://gvs.gs.washington.edu/SeattleSeqAnnotation/). For DP-specific, non-synonymous SNVs that result in missense mutations, we assessed the level of amino-acid conservation using PhyloP score (provided in UCSC genome database) where a score >2 implies high conservation and the nature of amino substitution using Polyphen-2 analysis18.
CNV analysis was performed using an R package, ExomeCNV15. ExomeCNV uses the ratio of read depth between two samples at each capture interval. Here, the read depth data between baseline and DP melanomas were compared. Briefly, the read depth information was extracted through the PILEUP file generated from the BAM file after removing PCR duplicates using SAMtools. The average read depth at each capture interval was calculated and the classify.eCNV module of ExomeCNV was run with the default parameters to calculate the copy-number estimate for each interval. Subsequently, another R package commonly used to segment the copy-number intervals, DNAcopy19, was called through ExomeCNV multi.CNV.analyze module with default parameters to do segmentation and sequential merging. The genomic regions with copy number 1 were called deletion and any regions with copy number >2 were called amplification. Circos20 was used to visualize the CNV data.
Protein detection
Western blots were probed with antibodies against p-ERK1/2 (T202/Y204), ERK1/2, C-RAF, AKT (Ser473), AKT (Thr308), AKT (Cell Signaling Technologies; all at 1:1,000), N-RAS, B-RAF (Santa Cruz Biotechnology; both at 1:500) and tubulin (Sigma; 1:700). For B-RAF immunohistochemistry, paraffin-embedded formalin-fixed tissue sections were antigen-retrieved, incubated with the primary antibody (Santa Cruz Biotechnology; 1:50) followed by HRP-conjugated secondary antibody (Envision System, DakoCytomation). Immunocomplexes were visualized using the DAB (3,3′-diaminobenzidine) peroxidase method and nuclei were counterstained with hematoxylin.
Genomic DNA and RNA quantifications
For real-time Q–PCR, total RNA was extracted and cDNA quantified by the iCycler iQ Real-Time PCR Detection System (Bio-Rad). Data were normalized to TUBULIN and GAPDH levels. Relative expression is calculated using the delta-Ct method. gDNAs were extracted using the FlexiGene DNA Kit (Qiagen; Human Genomic DNA-Female, Promega). B-RAF relative copy number was determined by Q–PCR (cycle conditions available upon request) using the MyiQ single colour Real-Time PCR Detection System. Total DNA content was estimated by assaying β-globin for each sample, and 20 ng of gDNA was mixed with the SYBR Green Q–PCR Master Mix (Bio-Rad) and 2 pmol l−1 of each primer. All primer sequences are provided in Supplementary Table S4.
Data processing
Statistical analyses were performed using InStat 3 Version 3.0b (GraphPad Software); graphical representations using DeltaGraph or Prism (Red Rock Software); and CI calculation using CalcuSyn V2.1 (Biosoft). Calculations were made by CalcuSyn software using the method of Chou and Taladay. Interpretation of CI values is summarized as follows: CI <0.1 (very strong synergy); 0.1–0.3 (strong synergy); 0.3–0.7 (synergy); 0.7–0.85 (moderate synergy); 0.85–0.9 (slight synergy); 0.90–1.10 (nearly additive); and 1.10–1.20 (slight antagonism). The relevant correlated Log10 (CI) values are shown as follow: Log10 (CI 0.1)=−1; Log10 (CI 0.3)=−0.5228787452803376; Log10 (CI 0.7)=−0.1549019599857432 and Log10 (CI 0.85)=−0.07058107428570727.
Additional information
Accession codes: Sequence data are archived at the NCBI Sequence Read Archive (SRA) under the accession code SRP010266.
How to cite this article: Shi, H. et al. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 3:724 doi: 10.1038/ncomms1727 (2012).
Accession codes
References
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002).
Bollag, G. et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599 (2010).
Chapman, P. B. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 (2011).
Flaherty, K. T. et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819 (2010).
Kefford, R. et al. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. J. Clin. Oncol. 28 (Suppl) abstr. 8503 (2010).
Ribas, A. et al. BRIM-2: an open-label, multicenter phase II study of vemurafenib in previously treated patients with BRAF V600E mutation-positive metastatic melanoma. J. Clin. Oncol. 29 (Suppl) abstr. 8509 (2011).
Shi, H., Kong, X., Ribas, A. & Lo, R. S. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to B-RAF(V600E) inhibition. Cancer Res. 71, 5067–5074 (2011).
Villanueva, J. et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18, 683–695 (2010).
Johannessen, C. M. et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 (2010).
Nazarian, R. et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010).
Poulikakos, P. I. et al. Acquired resistance to RAF inhibitors is mediated by splicing isoforms of BRAF (V600E) that dimerize in a RAS independent manner. Nature 480, 387–390 (2011).
Wagle, N. et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 29, 3085–3096 (2011).
Dumaz, N. et al. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 66, 9483–9491 (2006).
Infante, J. R. et al. Phase I/II study to assess safety, pharmacokinetics, and efficacy of the oral MEK 1/2 inhibitor GSK1120212 (GSK212) dosed in combination with the oral BRAF inhibitor GSK2118436 (GSK436). J. Clin. Oncol. 29 (Suppl) abstr. CRA8503 (2011).
Sathirapongsasuti, J. F. et al. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics 27, 2648–2654 (2011).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Olshen, A. B., Venkatraman, E. S., Lucito, R. & Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Acknowledgements
We are grateful to G. Bollag (Plexxikon Inc.) for providing PLX4032, J. S. Economou for biopsies, N. Doan for immunohistochemistry, B. Chmielowski and J. Glaspy for coordinated patient care, T.L. Toy for technical help with library generation for deep sequencing, and B. Harry for help with analysis of whole-exome sequence data. R.S.L. acknowledges funding from the following: Burroughs Wellcome Fund, National Cancer Institute (K22CA151638), V Foundation for Cancer Research, Melanoma Research Foundation, Melanoma Research Alliance, American Skin Association, Joint Center for Translational Medicine, Sidney Kimmel Foundation, Stand Up to Cancer, Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research, the Wesley Coyle Memorial Fund, Ian Copeland Melanoma Fund, Ruby Family Foundation, Louis Belley and Richard Schnarr Fund, and The Seaver Institute. R.F.K. and G.V.L. are supported by Program Grant No. 402761 from the National Health and Medical Research Council of Australia, Translational Research Program Grant No. 05/TPG/1-01 from the Cancer Institute New South Wales (CINSW). J.A.S. is supported by National Cancer Institute (K24CA097588) and American Cancer Society Melanoma Professorship. We are grateful to every patient volunteer who donated tissue(s) for this study.
Author information
Authors and Affiliations
Contributions
H.S., G.M., X.K., M.-K.L., H.L. designed, performed experiments and analysed data. R.C.K., C.N., T.C., R.A.S., K.B.D., J.A.S., R.F.K., G.V.L., A.R., and R.S.L. recruited patient volunteers and/or provided reagents/tissues. H.S., G.M., X.K, M.-K.L., H.L., J.A.S., R.F.K., G.V.L., S.F.N. and A.R. contributed to manuscript preparation. R.S.L. designed experiments and research aims, analysed data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
A.R. and R.S.L. are the authors of patent application under PCT Application Serial No. PCT/US11/061552 (Compositions and methods for detection and treatment of B-RAF inhibitor-resistant melanomas).
Supplementary information
Supplementary Information
Supplementary Figures S1-S6 and Supplementary Tables S1-S4 (PDF 964 kb)
Rights and permissions
About this article
Cite this article
Shi, H., Moriceau, G., Kong, X. et al. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 3, 724 (2012). https://doi.org/10.1038/ncomms1727
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms1727
This article is cited by
-
BRAF — a tumour-agnostic drug target with lineage-specific dependencies
Nature Reviews Clinical Oncology (2024)
-
Targeting metabolism by B-raf inhibitors and diclofenac restrains the viability of BRAF-mutated thyroid carcinomas with Hif-1α-mediated glycolytic phenotype
British Journal of Cancer (2023)
-
Towards precision oncology with patient-derived xenografts
Nature Reviews Clinical Oncology (2022)
-
Role of dual specificity phosphatases (DUSPs) in melanoma cellular plasticity and drug resistance
Scientific Reports (2022)
-
Genetic alteration of Chinese patients with rectal mucosal melanoma
BMC Cancer (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
