
Abstract
The oxidative pentose phosphate pathway (PPP) contributes to tumour growth, but the precise contribution of 6-phosphogluconate dehydrogenase (6PGD), the third enzyme in this pathway, to tumorigenesis remains unclear. We found that suppression of 6PGD decreased lipogenesis and RNA biosynthesis and elevated ROS levels in cancer cells, attenuating cell proliferation and tumour growth. 6PGD-mediated production of ribulose-5-phosphate (Ru-5-P) inhibits AMPK activation by disrupting the active LKB1 complex, thereby activating acetyl-CoA carboxylase 1 and lipogenesis. Ru-5-P and NADPH are thought to be precursors in RNA biosynthesis and lipogenesis, respectively; thus, our findings provide an additional link between the oxidative PPP and lipogenesis through Ru-5-P-dependent inhibition of LKB1–AMPK signalling. Moreover, we identified and developed 6PGD inhibitors, physcion and its derivative S3, that effectively inhibited 6PGD, cancer cell proliferation and tumour growth in nude mice xenografts without obvious toxicity, suggesting that 6PGD could be an anticancer target.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others


Accession codes
References
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Cairns, R. A., Harris, I. S. & Mak, T. W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 (2011).
Kroemer, G. & Pouyssegur, J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13, 472–482 (2008).
Christofk, H. R. et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233 (2008).
Hitosugi, T. et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22, 585–600 (2012).
Locasale, J. W. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 (2011).
Possemato, R. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 (2011).
Vander Heiden, M. G. et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329, 1492–1499 (2010).
Tian, W. N. et al. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem. 273, 10609–10617 (1998).
Farquharson, C., Milne, J. & Loveridge, N. Mitogenic action of insulin-like growth factor-I on human osteosarcoma MG-63 cells and rat osteoblasts maintained in situ: the role of glucose-6-phosphate dehydrogenase. Bone Miner. 22, 105–115 (1993).
Tian, W. N. et al. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am. J. Physiol. 276, C1121–C1131 (1999).
Li, D. et al. A new G6PD knockdown tumor-cell line with reduced proliferation and increased susceptibility to oxidative stress. Cancer Biother. Radiopharm. 24, 81–90 (2009).
Schafer, Z. T. et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 (2009).
Budihardjo, I. I. et al. 6-Aminonicotinamide sensitizes human tumor cell lines to cisplatin. Clin. Cancer Res. 4, 117–130 (1998).
Bravard, A., Luccioni, C., Muleris, M., Lefrancois, D. & Dutrillaux, B. Relationships between UMPK and PGD activities and deletions of chromosome 1p in colorectal cancers. Cancer Genet. Cytogenet. 56, 45–56 (1991).
Jonas, S. K. et al. Increased activity of 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase in purified cell suspensions and single cells from the uterine cervix in cervical intraepithelial neoplasia. Br. J. Cancer 66, 185–191 (1992).
Basu, J. et al. Alterations in erythrocyte glutathione metabolism associated with cervical dysplasias and carcinoma in situ. Cancer Invest. 11, 652–659 (1993).
Giusti, L. et al. Fine-needle aspiration of thyroid nodules: proteomic analysis to identify cancer biomarkers. J. Proteome Res. 7, 4079–4088 (2008).
Sukhatme, V. P. & Chan, B. Glycolytic cancer cells lacking 6-phosphogluconate dehydrogenase metabolize glucose to induce senescence. FEBS Lett. 586, 2389–2395 (2012).
Chan, B., VanderLaan, P. A. & Sukhatme, V. P. 6-Phosphogluconate dehydrogenase regulates tumor cell migration in vitro by regulating receptor tyrosine kinase c-Met. Biochem. Biophys. Res. Commun. 439, 247–251 (2013).
Shaw, R. J. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl Acad. Sci. USA 101, 3329–3335 (2004).
Woods, A. et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 (2003).
Shackelford, D. B. & Shaw, R. J. The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 (2009).
Park, S. H. et al. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J. Appl. Physiol. 92, 2475–2482 (2002).
Hardie, D. G. Regulation of fatty-acid and cholesterol-metabolism by the AMP-activated protein-kinase. Biochim. Biophys. Acta 1123, 231–238 (1992).
Jeon, S. M., Chandel, N. S. & Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012).
Fullerton, M. D. et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 19, 1649–1654 (2013).
Sommercorn, J. & Freedland, R. A. Regulation of hepatic phosphofructokinase by 6-phosphogluconate. J. Biol. Chem. 257, 9424–9428 (1982).
Davies, S. P., Sim, A. T. & Hardie, D. G. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur. J. Biochem. 187, 183–190 (1990).
Ha, J., Daniel, S., Broyles, S. S. & Kim, K. H. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J. Biol. Chem. 269, 22162–22168 (1994).
Kudo, N., Barr, A. J., Barr, R. L., Desai, S. & Lopaschuk, G. D. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 270, 17513–17520 (1995).
Munday, M. R., Campbell, D. G., Carling, D. & Hardie, D. G. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur. J. Biochem. 175, 331–338 (1988).
Mihaylova, M. M. & Shaw, R. J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 (2011).
Boudeau, J. et al. MO25α/β interact with STRADα/β enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 22, 5102–5114 (2003).
Zeqiraj, E., Filippi, B. M., Deak, M., Alessi, D. R. & van Aalten, D. M. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science 326, 1707–1711 (2009).
Marignani, P. A. et al. Novel splice isoforms of STRADα differentially affect LKB1 activity, complex assembly and subcellular localization. Cancer Biol. Ther. 6, 1627–1631 (2007).
Shan, C. et al. Lysine acetylation activates 6-phosphogluconate dehydrogenase to promote tumor growth. Mol. Cell 55, 552–565 (2014).
Fan, J. et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol. Cell. Biol. 31, 4938–4950 (2011).
Fan, J. et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 53, 534–548 (2014).
Hitosugi, T. et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 44, 864–877 (2011).
Hitosugi, T. et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2, ra73 (2009).
Hitosugi, T. et al. Tyr26 phosphorylation of PGAM1 provides a metabolic advantage to tumours by stabilizing the active conformation. Nat. Commun. 4, 1790 (2013).
Shan, C. et al. Tyr-94 phosphorylation inhibits pyruvate dehydrogenase phosphatase 1 and promotes tumor growth. J. Biol. Chem. 289, 21413–21422 (2014).
Yi, W. et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337, 975–980 (2012).
Yanagawa, T. et al. Differential regulation of phosphoglucose isomerase/autocrine motility factor activities by protein kinase CK2 phosphorylation. J. Biol. Chem. 280, 10419–10426 (2005).
Zhao, S. et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324, 261–265 (2009).
Lin, R. et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 51, 506–518 (2013).
Natali, F., Siculella, L., Salvati, S. & Gnoni, G. V. Oleic acid is a potent inhibitor of fatty acid and cholesterol synthesis in C6 glioma cells. J. Lipid Res. 48, 1966–1975 (2007).
Soh, Y., Song, B. J., Jeng, J. & Kallarakal, A. T. Critical role of arg433 in rat transketolase activity as probed by site-directed mutagenesis. Biochem. J. 333, 367–372 (1998).
Lee, J. Y., Cheong, D. E. & Kim, G. J. A novel assay system for the measurement of transketolase activity using xylulokinase from Saccharomyces cerevisiae. Biotechnol. Lett. 30, 899–904 (2008).
Wu, C. et al. Cordycepin activates AMP-activated protein kinase (AMPK) via interaction with the γ1 subunit. J. Cell. Mol. Med. 18, 293–304 (2014).
Acknowledgements
This work was supported in part by NIH grants CA140515, CA183594, CA174786 (J.C.), CA175316 (S.K.), GM071440 (C.H.) and the Pharmacological Sciences Training Grant T32 GM008602 (S.E.), DoD grant W81XWH-12-1-0217 (J.C.), National Natural Science Funds of China No. 20902013 (L.Zhou), Charles Harris Run For Leukemia, Inc. (H.J.K.) and the Hematology Tissue Bank of the Emory University School of Medicine and the Georgia Cancer Coalition (H.J.K.). T.H. is a Fellow Scholar of the American Society of Hematology. S.E. is a NIH pre-doctoral fellow and an ARCS Foundation Scholar. H.J.K., F.R.K., S.K. and J.C. are Georgia Cancer Coalition Distinguished Cancer Scholars. S.K. is a Robbins Scholar. S.K. and J.C. are American Cancer Society Basic Research Scholars. J.C. is a Scholar of the Leukemia and Lymphoma Society.
Author information
Authors and Affiliations
Contributions
R.L., S.E. and C.S. contributed equally to this work. J.X., T.-L.G., S.Z., K.Y., P.R.C., D.J.B., M.L.A., S.L., H.J.K., Q.L. and F.R.K. provided critical reagents. S.J.H. performed data analysis of pharmacokinetics studies. M.T. and T.-L.G. performed mass spectrometry-based assays. Q.J., L.Zhou, L.Zhang and C.H. performed biochemical analysis of lysine-acetylated 6PGD and molecular docking studies and analysed the data. J.S., L.J., M.M., R.J.D., S.W., Y.L. and H.M. performed quantitative mass spectrometry and NMR-based assays, and analysed data. B.H.L. performed the histopathological analyses. T.J.B. performed structural analyses. D.W. and G.Z.C. helped with xenograft experiments. C.S., S.E., H.-B.K., J.H.S., T.H. and J.F. performed all other experiments. R.L., S.E., C.S., S.K., J.F. and J.C. designed the study and wrote the paper. S.K., J.F. and J.C. are senior authors and jointly managed the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 6PGD is important for tumor growth and contributes to regulation of glycolysis.
(a) Tumor growth in xenograft nude mice injected with 6PGD KD (knockdown) K562 cells and control vector cells. (b) Left: Dissected tumors in a representative nude mouse and expression of 6PGD in tumor lysates. Scale bar represents 5 mm. Right: tumor mass in xenograft nude mice injected with 6PGD KD K562 cells and control vector cells. (c) 6PGD activity (upper) and expression (lower) in K562 cells with inducible knockdown of 6PGD in the presence and absence of Dox (doxycycline). (d) Cell proliferation rates determined by cell counting in K562 cells with induced 6PGD knockdown and control cells. (e) Tumor growth in xenograft nude mice injected with K562 cells with inducible 6PGD knockdown fed with drinking water (+/− Dox). (f) Dissected tumors in two representative nude mice (left) and tumor mass (right) of xenograft mice injected with K562 cells with inducible 6PGD knockdown fed with drinking water (+/− Dox). Scale bar represents 2 mm. (g) 6PGD activity (left) and protein expression (right) in tumor lysates from mice injected with K562 cells with inducible 6PGD knockdown fed with drinking water in the presence and absence of Dox are shown. (h–i) 6PGD KD H157 and K562 cells were tested for oxidative PPP flux (h) and NADPH/NADP+ ratio (i). (j) 6PGD KD K562 cells were tested for intracellular Ru-5-P (left) and R-5-P (right) levels. (k) 6PGD KD H157 and K562 cells were tested for RNA biosynthesis. (a,e) Mean ± S.E.M.; n = 8 tumors from 8 mice. (b,f) n = 8 tumors from 8 mice; centerlines represent means. The P values were determined by paired Student’s t-test for (b) and unpaired Student’s t-test for (a,e,f) (∗: 0.01 < p < 0.05; ∗∗: 0.001 < p < 0.01; ∗∗∗: p < 0.001). (a–d, g–k) Data are from a single experiment that is representative of 2 independent experiments for (a,b,d,g–k) and 3 independent experiments for (c). Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 2 6PGD controls Ru-5-P level to inhibit LKB1.
(a) K562 cells with inducible knockdown of 6PGD were tested for oxidative PPP flux (left) and lipogenesis (right) (+/− Dox). (b) 6PGD KD H1299 cells treated with Ru-5-P were tested for intracellular R-5-P levels. (c) Western blot results show AMPK knockdown by shRNA in 6PGD KD H1299 cells. (d) Lysates of 6PGD KD H1299 cells were treated with NADPH, followed by Western blot. (e) Lysates of 6PGD KD K562 cells were treated with Ru-5-P (left) or R-5-P (right), followed by Western blot. (f) Purified active AMPK proteins from H1299 cells were incubated with Ru-5-P, followed by in vitro AMPK kinase activity assay using SAMS peptide as substrate in the presence of γ-32P-ATP. The AMPK activity was assessed by incorporated amount of γ-32P in SAMS peptide and normalized to the control sample without Ru-5-P treatment. (g) Purified active LKB1 complex was incubated with recombinant AMPK in the presence of Ru-5-P, followed by Western blot to detect AMPK phosphorylation (p-T172). (h) Purified LKB1 complex was incubated with AMPK (upper) or MBP (lower) as substrates in the presence of NADPH, followed by Western blot to detect AMPK phosphorylation (upper) and Ser/Thr phosphorylation of MBP using a pan phospho-Ser/Thr antibody (lower). (i,j) Lysates from H1299 cells treated with Ru-5-P were applied to immunoprecipitation of LKB1 and Western blot to detect co-immunoprecipitated MO25 and STRAD (i), and protein expression levels of MO25, LKB1 and STRAD (j). (k) Lysates from H1299 cells treated with NADPH were applied to LKB1 immunoprecipitation and Western blot. (l) Cell lysates of 6PGD KD K562 cells were incubated with Ru-5-P (left) or R-5-P (right), followed by MO25 immunoprecipitation and Western blot. (m) Purified active LKB1 complex were incubated with R-5-P (left) or NADPH (right) for 20 minutes, followed by immunoprecipitation of LKB1 and Western blot. Data are from a single experiment that is representative of 2 independent experiments for (a,b,d,f,h–m) and 3 independent experiments for (c,e,g). Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 3 6PGD is important for regulation of lipogenesis and ROS levels in cancer cells.
(a) 6PGD KD H1299 cells were assayed for lipogenesis in the absence and presence of NAC (3 mM). The relative lipogenesis rates were normalized to the control vector cells without NAC treatment. (b) 6PGD KD 212LN cells were assayed for general ROS levels in the absence and presence of NAC by measuring ROS-mediated DCFDA oxidation to fluorescent DCF in the cells by flow cytometry. The relative general ROS levels were normalized to the control vector cells without NAC treatment. (c–e) Proliferation rates of H1299 cells treated with Compound C and/or NAC were determined using a cell number-based assay. (f) Proliferation rates of H1299 cells with AMPK knockdown by shRNA (left) were determined using a cell number-based assay. (g) Cell proliferation rates were determined by cell counting in 6PGD KD H1299 cells cultured in the absence and presence of NAC (left), Compound C (middle) or AMPK shRNA (right). Samples were prepared using same number of cells. (h) Effect of LKB1 knockdown (left) on AMPK phosphorylation in 6PGD knockdown cells was examined by Western blot. (i) H1299 cells with knockdown of 6PGD and/or LKB1 were tested for intracellular Ru-5-P levels (left) and lipogenesis (right). (j–n) 6PGD knockdown A549 cells with expression of LKB1 WT or the kinase-dead form K78M were tested for AMPK activation using Western blot (j), lipogenesis in the absence (k) or presence (l) of Ru-5-P, cell proliferation in the presence and absence of NAC treatment (m), and intracellular Ru-5-P levels (n). Data are from a single experiment that is representative of 2 independent experiments for (a,b,d,f,h–n) and 3 independent experiments for (c,e,g). Source data for independent replications and experiments with sample size <5 are available Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 4 Physcion and S3 are 6PGD inhibitors.
(a) K562 cells treated with increasing concentrations of Physcion were assayed for 6PGD (left) and G6PD (right) enzyme activity, which was normalized to the control cells without drug treatment. (b) Cell viability was determined in 6PGD KD K562 cells and control vector cells in the presence of increasing concentrations of Physcion. (c) Purified 6PGD WT (left) and M15A mutant (right) were assayed for 6PGD enzyme activity in the presence of increasing concentrations of S3. Absolute IC50 values are shown; NR = not reached. (d) H1299 6PGD knockdown cells were transiently transfected with vectors encoding 6PGD WT and M15A mutant, followed by Western blot. (e) H1299 6PGD knockdown cells were transiently transfected with vectors encoding 6PGD WT (left) and M15A mutant (right), followed by cell proliferation assay based on cell numbers in the presence of increasing concentrations of S3. Absolute IC50 values are shown; NR = not reached. (f) Cell proliferation rates of K562, MDA-MB-231 and 212LN cells in the presence of increasing concentrations of Physcion were determined by cell counting. (g) Apoptotic cell death of H1299 cells treated with increasing concentrations of Physcion for 12 hours, which was determined by annexin V staining. (h–l) K562 cells were assayed for intracellular concentration of 6-PG (h) and Ru-5-P (i), NADPH/NADP+ ratio (j), oxidative PPP flux and lipids (k), as well as apoptosis (l) in the presence and absence of Physcion. Data are from a single experiment that is representative of 2 independent experiments for (a–f,h–l) and 3 independent experiments for (g). Source data for independent replications and experiments with sample size <5 are available Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 5 6PGD inhibitors attenuate cancer cell proliferation.
(a,b) H1299 and K562 cells were assayed for intracellular 6-PG (a) and Ru-5-P (b) concentrations in the presence and absence of S3. (c) H1299 and K562 cells were treated with increasing concentrations of Physcion (left panels) or S3 (right panels), followed by PFK enzyme activity assay. (d,e) H1299 and K562 cells were treated with increasing concentrations of Physcion, followed by assays for lactate production (d) and intracellular ATP levels (e). (f) H1299 cells treated with or without S3 were assayed for cell proliferation rates by cell counting in the presence and absence of NAC. (g) K562 cells were treated with increasing concentrations of Physcion, followed by Western blot to detect phosphorylation levels of ACC1 (p79; upper) and AMPK (pT172; lower). 6PGD KD cells were included as a control. (h,i) K562 cells treated with or without Physcion were assayed for general ROS levels (h) and cell proliferation rates by cell counting (i) in the presence and absence of NAC. (j) K562 cells treated with or without Physcion were assayed for cell proliferation rates by cell counting in the presence and absence of Compound C (left) or lentivirus harboring AMPK shRNA (middle and right). (k–o) Effects of Physcion treatment on normal proliferating HFF cells were assayed for 6PGD activity (k), oxidative PPP flux rate (l), phosphorylation levels of AMPK (m) and lipid biosynthesis (n), as well as cell proliferation rates by cell counting (o). Data are from a single experiment that is representative of 2 independent experiments for (a,b,d,f,h–o) and 3 independent experiments for (c,e,g). Source data for independent replications and experiments with sample size <5 are available in SI Table 1. Uncropped Western blots are provided in Supplementary Information.
Supplementary Figure 6 S3 effectively inhibits tumor growth in xenograft nude mice.
(a) Cell viability of human cancer cells treated with S3 was determined by MTT assay. Normal proliferating human melanocyte PIG1 cells and human dermal fibroblasts (HDF) were included as controls. (b,c) Tumor growth (b) and tumor mass (c; left) in K562 cell-xenograft nude mice treated with S3 or DMSO. P values were determined by a two-tailed unpaired Student’s t test. c, right: Dissected tumors in representative nude mice treated with DMSO control or S3 are shown. Scale bar represents 2 mm. (d) 6PGD activity (upper) and protein expression (lower) in tumor lysates of K562 xenograft mice treated with DMSO or S3. (e) Representative images of IHC staining of Ki-67 (brown color) from K562 xenograft mice treated with DMSO or S3 are shown. Scale bars represent 50 μM. (f) 6PGD activity and cell proliferation in Tu212 cells treated with Physcion. (g) Dissected tumors in representative nude mice injected with Tu212 cells and treated with DMSO or S3. Scale bar represents 2 mm. (h,i) Effects of chronic treatment with S3 or DMSO on body weights (h) and serum chemistry (i) of nude mice are shown. ALT: alanine aminotransferase; AST: aspartate aminotransferase. (j) Peripheral blood samples from S3 or DMSO-treated H1299 xenograft nude mice (3 representative mice) were examined for hematological properties using CBC analysis. (k) Histological morphology of hematoxylin-eosin stained tissue sections of representative H1299 xenograft nude mice in S3 or DMSO-treated groups presented in Figure 8a–8d. Images were analyzed and captured using ImageScope software (Aperio Technologies Inc.) without any additional or subsequent image processing (high power images are 20×; low power images are 4×). Scale bars are indicated. (b) Mean ± S.E.M.; n = 6 tumors from 6 mice. (c,h) n = 6 tumors from 6 mice; (c) centerlines represent means. (i,j) Mean ± S.D.; n = 3 serum from 3 mice. P values were obtained by two-sided Student’s t-test for (b,c) (∗: 0.01 < p < 0.05). (a–d,f) Results of one representative experiment from two independent experiments are shown. Source data for independent replications and experiments with sample size <5 are available Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 7 Physcion and S3 effectively inhibit cell proliferation of human primary leukemia cells from human patients with minimal toxicity.
(a) Effect of treatment with Physcion on cell proliferation was examined in human primary leukemia cells isolated from bone marrow (BM) samples from a representative AML patient. (b,c) Effect of Physcion treatment on lactate production (b) and intracellular ATP levels (c) in human primary leukemia cells isolated from PB samples from a representative CML patient. (d) Effect of S3 treatment on cell viability was examined in human primary leukemia cells isolated from PB samples from a representative B-ALL patient. (e,f) Effect of treatment with S3 on cell proliferation of human primary leukemia cells isolated from PB samples from a representative B-ALL patient (e) and cell viability human primary leukemia cells isolated from PB or BM samples from two AML and one CML patients (f) were examined. (g) S3 shows no toxicity in treatment (72h) of peripheral blood cells from a representative healthy human donor. (h) Physcion and S3 show no toxicity in treatment (72h) of CD34+ cells isolated from bone marrow samples from representative healthy human donors. (a–h) The data are obtained from peripheral blood or bone marrow of multiple human primary tissue samples. Each sample was used for a single experiment of 3 technical replicates. Source data for experiments with sample size <5 are available in Supplementary Table 1.
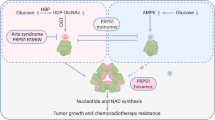
Supplementary Figure 8 6PGD provides an additional link between oxidative PPP and lipogenesis.
(a) Proposed working model. Left: In cancer cells, 6PGD activity is commonly upregulated by lysine acetylation, which promotes oxidative PPP and subsequent biosynthesis of nucleotide and RNA. The intracellular Ru-5-P is maintained at a physiological level that is sufficient to inhibit LKB1-AMPK signaling, leading to relief of AMPK-dependent inhibition of ACC1 and consequently high levels of lipid biosynthesis to fulfill the request of rapidly growing tumors. Right: Attenuation of 6PGD by shRNA or small molecule inhibitors results in decreased Ru-5-P levels, leading to reduced nucleotide/RNA biosynthesis and increased LKB1-AMPK signaling, which inhibits ACC1 and lipogenesis. (b,c) H1299 cells with stable knockdown of H6PD (b) or G6PD (c) were tested for AMPK phosphorylation (left), Ru-5-P levels (middle), and cell proliferation (right). Data are from a single experiment that is representative of 2 independent experiments for (b) and 3 independent experiments for (c). Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary information
Supplementary Information
Supplementary Information (PDF 1393 kb)
Supplementary Table 1
Supplementary Information (XLSX 324 kb)
Supplementary Table 2
Supplementary Information (XLS 30 kb)
Supplementary Table 3
Supplementary Information (XLS 24 kb)
Rights and permissions
About this article

Cite this article
Lin, R., Elf, S., Shan, C. et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1–AMPK signalling. Nat Cell Biol 17, 1484–1496 (2015). https://doi.org/10.1038/ncb3255
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncb3255
This article is cited by
-
Mangiferin alleviates diabetic pulmonary fibrosis in mice via inhibiting endothelial-mesenchymal transition through AMPK/FoxO3/SIRT3 axis
Acta Pharmacologica Sinica (2024)
-
Metabolic reprogramming in colorectal cancer: regulatory networks and therapy
Cell & Bioscience (2023)
-
New insights into activation and function of the AMPK
Nature Reviews Molecular Cell Biology (2023)
-
N6-methyladenosine modification of circ_0003215 suppresses the pentose phosphate pathway and malignancy of colorectal cancer through the miR-663b/DLG4/G6PD axis
Cell Death & Disease (2022)
-
Mechanosensitive turnover of phosphoribosyl pyrophosphate synthetases regulates nucleotide metabolism
Cell Death & Differentiation (2022)
