
Abstract
During apoptosis, Bak and Bax permeabilize the mitochondrial outer membrane by undergoing major conformational change and oligomerization. This activation process in Bak is reported to require dephosphorylation of tyrosine-108 close to an activation trigger site. To investigate how dephosphorylation of Bak contributes to its activation and conformational change, one-dimensional isoelectric focusing (1D-IEF) and mutagenesis was used to monitor Bak phosphorylation. On 1D-IEF, Bak extracted from a range of cell types migrated as a single band near the predicted isoelectric point of 5.6 both before and after phosphatase treatment, indicating that Bak is not significantly phosphorylated at any residue. In contrast, three engineered ‘phosphotagged’ Bak variants showed a second band at lower pI, indicating phosphorylation. Apoptosis induced by several stimuli failed to alter Bak pI, indicating little change in phosphorylation status. In addition, alanine substitution of tyrosine-108 and other putative phosphorylation sites failed to enhance Bak activation or pro-apoptotic function. In summary, Bak is not significantly phosphorylated at any residue, and Bak activation during apoptosis does not require dephosphorylation.
Similar content being viewed by others


Main
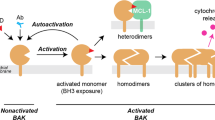
The mitochondrial pathway of apoptosis is regulated principally by the Bcl-2 protein family.1, 2 Each family member contains up to four Bcl-2 homology (BH) domains. The BH3-only members such as Bid and Bim contain just one BH domain, and act as triggers of apoptosis. Two other pro-apoptotic proteins, Bak and Bax, contain all four BH domains, and participate in the key step of pore formation in the mitochondrial outer membrane.3, 4 The pro-survival proteins, including Bcl-2 and Mcl-1, also contain all four BH domains, and act by sequestering both the BH3-only proteins and activated Bak and Bax.5, 6, 7 Interactions between family members involve binding of the BH3 domain of pro-apoptotic proteins to a surface hydrophobic groove, with sequence differences determining binding affinity.8, 9 Thus, cell death is regulated by the relative levels of Bcl-2 proteins, and by specific binding between members.
Non-activated Bak and Bax contain nine α-helices, with the hydrophobic α5-helix surrounded by amphipathic helices. During apoptosis, Bak and Bax activation involves major conformational change including exposure of N-terminal epitopes and the BH3 domain (α2), with the latter able to bind to the hydrophobic groove (α3–α5) of another activated molecule to generate symmetric homodimers.10, 11, 12 These symmetric dimers are thought to multimerize to form the high order oligomers that permeabilize mitochondria, perhaps via self-association of the α6-helices.13 Activation of Bak and Bax is triggered by binding of the ‘activator’ BH3-only proteins (e.g. Bid and Bim) to the hydrophobic groove in Bak,9 and perhaps the α1/α6 region in Bax.14 Thus, the hydrophobic groove is a critical site for Bak activation as well as its homodimerization.
Phosphorylation of several Bcl-2 family proteins regulates their apoptotic function (reviewed in15). For example, phosphorylation-mediated ubiquitination accounts in part for the short half-life of Mcl-1.16, 17, 18 Bim half-life is increased by phosphorylation at certain residues,19 but decreased by phosphorylation elsewhere.20 Phosphorylation of Bax at several residues has been linked to altered apoptotic function including altered mitochondrial translocation.15
Evidence of Bak phosphorylation derives from studies performed by two groups. In early studies, multiple Bak species following two-dimensional electrophoresis suggested multiple post-translational modifications, although the role of phosphorylation was not investigated.21 More recently, two-dimensional electrophoresis also showed multiple Bak species, with phosphatase treatment increasing the pI to that predicted (5.6) for Bak.22, 23 In addition, Bak shifted to a higher pI after apoptotic signaling, suggesting that Bak was dephosphorylated during apoptosis. Analysis of the Bak α4 region by mass spectroscopy indicated phosphorylation at four sites (Y108, Y110, T116 and S117). Notably, Y108 dephosphorylation appeared necessary for Bak activation and conformational change, as alanine substitution at this position increased Bak activation and pro-apoptotic function.22, 23
Given that α4 forms part of the canonical hydrophobic groove, a region implicated in both the activation and homo-oligomerization of Bak,9, 10 we explored how phosphorylation alters Bak function. Although one-dimensional isoelectric focusing (1D-IEF) readily detected phosphorylation of ‘phosphotagged’ Bak, phosphorylation of untagged, wild-type Bak (wtBak) was not evident in any cell type or condition examined. In addition, Bak pro-apoptotic function was unchanged by alanine or cysteine substitution of Y108, Y110, T116 or S117. These findings strongly argue that dephosphorylation of Bak is not required for its activation or function.
Results
1D-IEF to assess Bak phosphorylation
Bak phosphorylation has previously been analyzed by two-dimensional gel electrophoresis.21, 23 In this technique, the first dimension is isoelectric focusing (IEF) of a single sample to separate proteins on the basis of charge, and the second dimension is SDS-PAGE to separate on the basis of molecular weight. Here we used one-dimensional IEF as it allowed comparison of multiple Bak samples in the same gel.24, 25
As found for most membrane proteins,26 IEF of Bak was initially problematic, and required significant optimization of detergents and other buffer components to efficiently extract Bak from the mitochondrial outer membrane, and to focus the protein in the IEF gel. Once achieved however, human Bak (hBak) expressed in Bak−/−Bax−/− mouse embryonic fibroblasts (MEFs) routinely ran as a single species that focused near the predicted pI of 5.6 (Figure 1, lane 2). This pI reflects that of Bak in intact cells, as the same band was evident when membranes were prepared in the presence of a cocktail of phosphatase inhibitors, or only the tyrosine phosphatase inhibitor, sodium orthovanadate (Figure 1, lanes 3 and 4). Bak pI was also unaltered by incubation with λ-phosphatase to dephosphorylate all serine, threonine and tyrosine residues (Figure 1, lane 5). The IEF conditions could detect added negative charges as labeling with 4-acetamido-4′-[(iodoacetyl)amino]stilbene-2,2′-disulfonic acid (IASD) lowered Bak pI (Figure 1, lane 6). IASD contains two negative charges, and therefore imparts two or four negative charges when bound to one or both cysteines in Bak. These results suggest that Bak is not significantly phosphorylated in these cells.

Bak runs as a single band at the predicted isoelectric point on IEF. Membrane fractions from Bak−/−Bax−/− MEFs or those cells expressing wild-type hBak were run on one-dimensional IEF gels (pH 3–7) and immunoblotted for Bak. Where indicated, samples were prepared in the presence of a mixture of phosphatase inhibitors (J; β-glycerophosphate, sodium molybdate, sodium pyrophosphate, sodium fluoride and sodium orthovanadate), or a single tyrosine phosphatase inhibitor (N; sodium orthovanadate). Also where indicated, samples were incubated with λ-phosphatase (λ-ppase) or the sulfhydryl labeling agent, IASD. Data are representative of three independent experiments
To directly verify that our IEF conditions could detect Bak phosphorylation, we generated three Bak variants with N-terminal ‘phosphotags’ containing the protein kinase A (PKA) phosphorylation motif R/KR/KXpS/pT (Figure 2a). In phospho1Bak, the N-terminal tag contains five residues (RRASL) based on BimEL sequence (80RRSSL84) that is constitutively phosphorylated by PKA in MEFs.19 As Ser83 is the target of PKA, Ser82 was substituted for alanine to ensure a single phosphorylation target. As a control for no phosphorylation within this tag, Ser83 was replaced with alanine (phospho1S→ABak). A second variant, phospho2Bak, was based on a sequence (126EILSRRPSYR135) from the CREB (cAMP response element binding) protein. This tag had the potential to be phosphorylated at two serine residues, as phosphorylation of the second serine by PKA converts the first serine into a substrate of glycogen synthase kinase-3.27 A third variant, phospho3Bak, contains the HA-tag (DYPYDVPDYA) together with a linking sequence (TRR) that generates the RRASG sequence. Thus, all three phosphotagged Bak variants contained the PKA phosphorylation motif at the N-terminus, and had the potential to be serine phosphorylated at one or two positions.

Phosphotagged Bak variants confirm that IEF conditions can detect phosphorylated Bak. (a) Three tags containing the PKA phosphorylation motif (R/KR/KXpS/pT, underlined) were added to the Bak N-terminus to generate positive controls of Bak phosphorylation. phospho1S→ABak, which lacks serine in the tag, was generated as a negative control. Phosphorylatable serine residues are indicated (bold). pI and MW were calculated at http://web.expasy.org/protparam/, with a serine to glutamate substitution representing phosphorylation. (b) IEF detects phosphorylated Bak variants. Membrane fractions from Bak−/−Bax−/− MEFs expressing the indicated phosphotagged Bak variants were prepared in the presence of sodium orthovanadate, then resuspended in wash buffer, and incubated with or without λ-phosphatase before running on IEF and immunoblotting for Bak. *Phosphorylated species. Note that a lower band in the phospho1S→ABak samples was not evident in other experiments. (c) SDS-PAGE detects phosphorylation of phospho3Bak. Samples prepared for IEF in (b) were supplemented with SDS sample buffer and run on reducing SDS-PAGE before immunoblotting for Bak. Data are representative of three independent experiments
Each phosphotagged Bak variant was stably expressed in Bak−/−Bax−/− MEFs, and shown to retain pro-apoptotic function (data not shown). IEF of mitochondria-enriched membrane fractions showed two bands in each phosphotagged variant (Figure 2b). Upper bands focused to the pI of non-phosphorylated variants, whereas the lower bands represented the phosphorylated forms, as they were lost following λ-phosphatase treatment and were not present in the phospho1S→ABak sample. Only one serine in the tag of phospho2Bak appeared to be phosphorylated. In summary, a serine in each tag was efficiently phosphorylated in MEFs. More importantly, the ability to detect Bak with a single modification confirms that wild-type hBak stably expressed in MEFs is not significantly phosphorylated.
On SDS-PAGE, the phosphotagged Bak variants displayed aberrant migration (Figure 2c and Supplementary Figure 1). Firstly, phosphorylation caused slow migration of phospho3Bak, but not the other phosphotagged Bak variants (Figure 2c). This is occasionally observed (e.g. Bim19), despite little contribution of a phosphate group to the molecular weight. Secondly, the phosphotagged Bak variants (except for phosphorylated phospho3Bak) ran at a lower molecular weight than wtBak, despite 5–12 additional residues (Figure 2c). This faster migration correlated with the presence of two positively charged residues (RR) in each tag (Supplementary Figure 1). Differences in SDS binding may underlie both the slow (negative PO4 group) and fast (positive RR residues) migration of these Bak variants on SDS-PAGE.28
The phosphotagged Bak variants also helped to calibrate the pI gradient of IEF gels (Supplementary Figure 2a), and to show that phosphatase inhibitors present during Bak extraction were effective in blocking dephosphorylation (Supplementary Figure 2b).
Bak phosphorylation is not evident in a variety of cell lines
We next investigated whether endogenous Bak expressed in cells other than Bak−/−Bax−/− MEFs might be phosphorylated, as reported for HT1080 cells and hBak expressed in Bak−/− MEFs23 and perhaps Jurkat cells.21 Membrane fractions from a range of human and mouse cell lines were solubilized and assessed by IEF, but failed to show phosphorylation of either human or mouse Bak (Figure 3). Mouse Bak extracted from mouse-liver mitochondria or from primary MEFs also migrated as a single band before and after phosphatase treatment (Supplementary Figure 3). Thus, Bak is not significantly phosphorylated in either transformed or non-transformed cells.

Bak phosphorylation is not evident in a panel of human and mouse cells. Membrane fractions from the indicated cell lines were incubated with or without λ-phosphatase, run on IEF and immunoblotted for Bak. Note that hBak in lanes 3 and 4 focused unevenly due to non-uniform isoelectric gradient sometimes observed in these gels, but focused at the same pI in a repeat gel. Mouse Bak predicted pI is 5.79. *Phosphorylated species. Data are representative of three independent experiments
Bak phosphorylation status does not change during apoptosis
When analyzed by two-dimensional gel electrophoresis, Bak pI increased significantly when HT1080 cells underwent apoptosis induced by camptothecin or UV,23 although not during Jurkat-cell apoptosis induced by staurosporine.21 Thus we examined whether our IEF conditions would detect a change in Bak pI during apoptosis. MEFs were incubated with etoposide, staurosporine or camptothecin for 24 h, at which time around 50% of Bak was activated and formed homo-oligomers as shown by cysteine linkage (Figure 4a). However, no change in Bak pI was apparent (Figure 4b), indicating that Bak phosphorylation status does not change significantly during apoptosis. UV-treatment of MEFs also failed to alter Bak pI (data not shown).

Bak phosphorylation does not change during apoptosis. (a) Bak activation and oligomerization following apoptosis. Bak−/−Bax−/− MEFs expressing wild-type hBak were incubated with etoposide (10 μM), staurosporine (2.5 μM) or camptothecin (6 μM) for 24 h in the presence of the caspase inhibitor Q-VD-OPh. Membrane fractions were then incubated with oxidant (CuPhe) to induce disulfide linkage, and run on non-reducing SDS-PAGE before immunoblotting for Bak to reveal monomers (M), intramolecular disulfide-linked monomers (Mx), dimers (D) and trimers (T). (b) Bak pI does not change during apoptosis. Cells were treated as in (a), and membrane fractions analyzed by IEF and immunoblotted for Bak. As a positive control for Bak phosphorylation, lanes 1 and 2 contain membrane fractions from cells expressing phospho1Bak treated with or without λ-phosphatase. *Phosphorylated species. Data are representative of three independent experiments
Bak is not recognized by antibodies to phosphotyrosine
We next examined tyrosine phosphorylation specifically, as phosphorylation of Bak at Y108 was detected by mass spectroscopy, and proposed to block Bak activation.22, 23 After enriching for Bak by immunoprecipitation, even under cell-culture conditions where tyrosine phosphatases were blocked by sodium pervanadate, pTyr-containing Bak was not detected by one antibody to phosphotyrosine (Figure 5).

Bak tyrosine phosphorylation is undetectable even after inhibition of tyrosine phosphatases in cell culture. Bak−/−Bax−/− MEFs expressing wild-type hBak were treated with sodium pervanadate (H6Na3VO10), an inhibitor of tyrosine phosphatases, for 20 or 40 min. Membrane fractions were then prepared in the presence of phosphatase inhibitor cocktail, washed and incubated with or withoutλ-phosphatase prior to its lysis in 1% digitonin. Aliquots were immunoprecipitated for Bak and run alongside lysates on SDS-PAGE and immunoblotted with anti-phosphotyrosine antibody (top), then reblotted for Bak (bottom). A 50-kD band in immunoprecipitated samples is IgG heavy chain. Data are representative of three independent experiments
Non-phosphorylatable Bak retains pro-apoptotic function
Mutagenesis was then used to specifically address the role of putative Bak phosphorylation sites. Mass spectrometry of the Bak α4 region had shown phosphorylation not only at Y108, but at Y110, T116 and S117.23 As these residues are located in or near the hydrophobic groove (Figure 6a), their phosphorylation may alter binding and/or activation by BH3-only proteins. We thus substituted alanine at Y108, T116 or S117. We had previously substituted Y110 with the non-phosphorylatable cysteine and found no change in pro-apoptotic function.10 When stably expressed in Bak−/−Bax−/− MEFs, each alanine-substituted Bak was first assessed by IEF for an increase in pI (Figure 6b). However, each variant migrated as a single band at the predicted pI (same as wtBak), verifying that these specific sites are not phosphorylated in Bak.

Mutagenesis of putative phosphorylation sites in the α4-helix does not alter Bak function. (a) Four putative phosphorylation sites are positioned near the hydrophobic groove of Bak. Cartoon representation of non-activated Bak (2IMT;36) with α2–α4 region of the groove highlighted (black). Sidechains of four putative phosphorylated residues are shown. (b) Alanine substitution in α4 does not alter Bak pI. Membrane fractions from Bak−/−Bax−/− MEFs expressing the indicated Bak variants were treated with or without λ-phosphatase and run on IEF prior to immunoblotting for Bak. Data are representative of three independent experiments. (c) Alanine substitution at Y108, T116 or S117 does not alter Bak function. Cells in (b) were treated with etoposide or camptothecin for 24 h, and assessed for cell death (propidium iodide uptake). Bak levels prior to apoptosis are also shown (insert). Cell death (%) is expressed as mean±S.D. of three independent experiments
When treated with increasing doses of etoposide or campothecin, BakY108A showed similar pro-apoptotic function to wtBak (Figure 6c), in contrast to previous findings.23 Likewise, BakT116A showed similar pro-apoptotic function to wtBak, whereas BakS117A appeared slightly less functional, at least in response to camptothecin (Figure 6c). Whereas decreased function of BakS117A cannot be attributed to changes in phosphorylation (e.g. Figure 6b), serine substitution may alter Bak stability and propensity to undergo conformational change. Accordingly, S117 substitution with glutamate (initially generated to mimic phosphorylation) resulted in very low protein levels (data not shown).
Time-course experiments and Bak-activation assays further addressed whether BakY108A was more susceptible to activation as proposed.23 However, BakY108 and wtBak showed equal onset of apoptosis following etoposide, camptothecin or staurosporine (Figure 7a), including the use of polyclonal cell lines derived by retroviral transduction on three occasions (data not shown). In addition, conversion of BakY108A to the activated conformation was not greater than that of wtBak when assessed by exposure of the Ab-1 N-terminal epitope (Figure 7b). These mutagenesis studies confirm that phosphorylation at Y108 (or Y110, S116 and T117) does not have an impact on the ability of Bak to be activated or to mediate apoptosis.

BakY108A is not more readily activated than wtBak. (a) Onset of BakY108A-mediated apoptosis is equivalent to that of wtBak. Bak−/−Bax−/− MEFs expressing wt or Y108A Bak were treated with etoposide (10 μM), camptothecin (6 μM) or staurosporine (12.5 μM), and tested at the indicated time points for propidium iodide uptake. Cell death (%) is expressed as mean±S.D. of three independent experiments. (b) Exposure of the Ab-1 epitope in BakY108A following apoptosis is not more rapid than for wtBak. Cells treated for 24 h as in (a) and also with UV, in the presence of the caspase inhibitor Q-VD-OPh, were analyzed for Ab-1 exposure by flow cytometry. Cell detection with Ab-1 antibody (solid lines) or with secondary antibody only (broken lines) is shown. Cells (%) with Ab-1 exposure (to the right of marker) are indicated. Data are representative of three independent experiments
Discussion
Bak and Bax are the key apoptotic effectors. Hence, the molecular mechanisms by which they become activated are of intense interest due to the therapeutic benefits of altering Bak and Bax activation.1, 29 We have reported that a key step in Bak (and Bax) apoptotic function involves reciprocal interaction of the BH3 domain with the canonical hydrophobic groove of a partner molecule to form homodimers.10, 11, 30 The hydrophobic groove is also a binding site for direct activator BH3-only proteins Bid, Bim and Noxa.9 Therefore, reports that Y108 in the Bak groove is constitutively phosphorylated, and that dephosphorylation is a requisite event for Bak activation,22, 23 prompted us to examine the mechanism involved.
Using 1D-IEF, immunoprecipitation and mutagenesis, we found no evidence of Bak phosphorylation in several mouse and human cell lines, including after apoptosis. As blockade of Bak-mediated apoptosis by phosphorylation (or by any other means), would require a significant portion of the protein to be modified, our results argue that Bak activation does not require dephosphorylation at any residue. Our findings do not exclude cell-specific or context-specific roles for Bak phosphorylation, however, cells reported to support multiple Bak phosphorylation21, 23 did not do so when assessed by IEF conditions that readily detected phosphorylation of three phosphotagged Bak variants.
Our IEF conditions differ significantly from those previously used for Bak.21, 23 First, we found that the detergent ASB-16 and thiourea were both critical for the efficient extraction and isoelectric focusing of Bak, with other detergents including ASB-14, digitonin and TritonX-100 resulting in poor focusing. Second, the use of 1D-IEF allowed direct comparison of several Bak samples in each gel, controlling for variation in sample preparation and gel integrity. The current IEF approach could be used to examine post-translational modifications, including phosphorylation, of other membrane proteins, including other Bcl-2 family members.
Mutation of three putative phosphorylation sites in the Bak α4-helix also failed to support a role for phosphorylation within the hydrophobic binding groove in regulating Bak apoptotic function. In particular, substitution of Y108 with alanine did not alter Bak activation, oligomerization or apoptotic function. This contrasts with reports that BakY108A was more readily activated than wtBak.22, 23 Whereas the reasons for this discrepancy are not clear, in those studies BakY108A function was tested in Bak−/−MEFs and HCT116 cells that express Bax. As Bak and Bax can associate during apoptosis,11 the Y108A mutation may increase Bak binding to Bax to facilitate apoptosis. However, it is clear that in the absence of Bax, BakY108A does not kill more efficiently than wtBak (Figures 6 and 7).
Our studies have not examined whether Bak phosphorylation contributes to its degradation and turnover, as reported for several other Bcl-2-family members.15 Given the relatively long half-life of Bak (>24 h in MEFs),31 only a small portion of Bak may be phosphorylated at any one time, and thus not detected on IEF. Further analysis is required to determine whether phosphorylation of Bak, perhaps at Y108 and other sites identified by mass spectrometry,23 are important for Bak-protein turnover.
Whereas our results show that Bak apoptotic function is not directly regulated by phosphorylation, multiple targets upstream of Bak may be regulated by phosphorylation, and thus account for the ability of MEK/ERK signaling and PTPN5 to regulate Bak-mediated apoptosis.22, 23 Bim and Mcl-1 are just two candidates, as they both regulate Bak, and both are regulated by phosphorylation.32 Careful examination of such pathways may well identify important therapeutic targets.
Materials and Methods
Cell culture
MEFs were generated from C57BL/6 Bak−/−Bax−/− mouse embryos and transformed with SV40 large T as described.33 MEFs and HT1080 (human fibrosarcoma) cell lines were maintained in Dulbecco’s Modified Eagles medium (DMEM) supplemented with 10% fetal calf serum (FCS), 250 μM L-asparagine and 55 μM 2-mercaptoethanol. HeLa (human epithelial) and HCT116 (human colorectal carcinoma) cells were maintained in DMEM supplemented with 10% FCS. WEHI-231 (human B lymphocyte) cells were maintained in DMEM KELSO supplemented with 10% FCS, 1 mM L-asparagine, 55 μM 2-mercaptoethanol, 1 × non-essential amino acids and 1 mM L-glutamine.
Bak constructs and retroviral expression
N-terminal-phosphotagged Bak variants were generated by PCR mutagenesis, and alanine substitution of putative phosphorylated sites performed by site-directed mutagenesis and overlap extension PCR (primer sequences available on request). Bak variants were then stably expressed in Bak−/−Bax−/− MEFs by transduction with pMX-IG (IRES-GFP) retroviral constructs, and GFP-positive cells sorted to obtain polyclonal populations.10
Apoptosis assays
Apoptosis was initiated by exposure of cells to UV (100 J/m2) or by addition of the indicated concentrations of etoposide, staurosporine (Sigma-Aldrich, St Louis, MO, USA) or camptothecin (Calbiochem, Merck KGaA, Darmstadt, Germany). Cell death after 24-h incubation (unless indicated otherwise) was assessed by propidium iodide uptake using flow cytometry.10
Preparation of mitochondrial fractions using phosphatase and inhibitor treatments
To prepare membrane fractions enriched for mitochondria, cells were resuspended (1 × 107 cells/ml) in permeabilization buffer (250 mM sucrose, 20 mM HEPES, pH 7.5, 50 mM KCl, 2.5 mM MgCl2, 0.025% digitonin (Calbiochem), Complete protease inhibitor cocktail (Roche, Basel, Switzerland) and 4 μg/ml pepstatin A (Sigma-Aldrich)), and incubated on ice for 10 min. Permeabilized cells were then centrifuged at 13 000 × g for 5 min to obtain the membrane fraction containing mitochondria.
To block phosphatases in membrane fractions, permeabilization buffer was supplemented with either 2 mM activated sodium orthovanadate (Na3VO4) alone, or a cocktail of phosphatase inhibitors (5 mM β–glycerophosphate, 1 mM sodium molybdate, 2 mM sodium pyrophosphate, 10 mM sodium fluoride and 2 mM activated Na3VO4 (each from Sigma-Aldrich)). Activated Na3VO4 was prepared as previously described.34 Briefly, the Na3VO4 solution (200 mM in water) was adjusted to pH 10 and boiled until colorless. After cooling to room temperature, the process was repeated until the solution remained colorless and the pH had stabilized at 10. Aliquots were stored at −20°C.
To inhibit tyrosine phosphatases during cell culture, sodium pervanadate (H6Na3VO10), which is cell permeable, was first generated by combining activated Na3VO4 (200 mM) with an equal volume of hydrogen peroxide (0.4 M) and incubating at room temperature for 10 min. The resulting sodium pervanadate was immediately added to cells at 1 mM.34, 35
To dephosphorylate at all serine, threonine and tyrosine residues, membrane fractions from permeabilized cells were resuspended in wash buffer (300 mM sucrose, 10 mM Tris-HCl pH 7.4, 1 mM EDTA, Complete protease inhibitors, and 4 μg/ml pepstatin A) to remove the majority of the phosphatase inhibitors, and treated with λ-phosphatase (New England Biolabs, Ipswich, MA, USA) at 30 °C for 30 min according to the manufacturer’s protocol. The reaction was stopped by adding either IEF or SDS-PAGE sample buffer.
SDS-PAGE and immunoblotting
SDS-PAGE was performed using 4–20 or 12% pre-cast Tris-glycine gels (Bio-Rad, Hercules, CA, USA). Samples were loaded according to equal cell number and transferred to polyvinylidene fluoride (PVDF) membranes. Bak was detected using rabbit polyclonal antibody (B5897, Sigma). Phosphorylated tyrosine was detected with an anti-pTyr antibody (PY99, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Secondary anti-rabbit and anti-mouse horseradish peroxidase-conjugated antibodies (Southern Biotech, Birmingham, AL, USA) were detected using Luminata Forte Western HRP substrate (Millipore, Billerica, MA, USA) and digital imaging (BioRad ChemiDoc XRS+ System).
1D-IEF
Membrane fractions prepared in permeabilization buffer supplemented with phosphatase inhibitor cocktail were resuspended (1 × 107 cells/ml) in wash buffer, and where indicated by treating with λ-phosphatase. Samples were then solubilized by addition of 1% ASB-16 (w/v; Merck, Darmstadt, Germany), incubated for 30 min on ice and spun at 13 000 × g for 5 min at 4 °C to remove unsolubilized cell debris. The supernatant was combined with an equal volume of IEF sample buffer (7 M urea, 2 M thiourea, 2% CHAPS, complete protease inhibitor, 4 μg/ml pepstatin A, 50 mM DTT, 1% ASB-16 and 0.04% bromophenol blue), and 25 μl immediately loaded onto Novex, pH 3–7 IEF gels (Invitrogen, Carlsbad, CA, USA). Gels were focused with increasing voltage (100 V for 1 h, 200 V for 1 h, 500 V for 30 min) powered by the Consort EV265 power pack (Consort, Turnhout, Belgium). Gels were then soaked for 5 min in SDS buffer (75 mM Tris/HCl, pH 6.8, 0.6% SDS, 15% glycerol) and transfered at 40 mA for 2.5 h to PVDF membranes, and immunoblotted as for SDS-PAGE. Samples prepared for IEF were sometimes also run on SDS-PAGE after addition of an equal volume of SDS sample buffer.
Detecting Bak activation and oligomerization
Bak activation and oligomerization was monitored by disulfide linkage between endogenous cysteines (C14 and C166) in hBak, as previously described.10 Briefly, membrane fractions were incubated with the oxidant copper(II)(1,10-phenanthroline)3 (CuPhe) on ice for 30 min before quenching the reaction with 20 mM EDTA, and then run on non-reducing SDS-PAGE. Activated Bak was also detected by exposure of the Ab-1 epitope as previously described.10
Bak immunoprecipitation
To assess Bak tyrosine phosphorylation in sodium pervanadate-treated MEFs, membrane fractions prepared in permeabilization buffer supplemented with phosphatase inhibitor cocktail were resuspended in permeabilization buffer containing 1% digitonin and incubated on ice for 30 min to solubilize Bak. The resulting lysate was immunoprecipitated using the 7D10 anti-Bak antibody that recognizes all forms of hBak.10
Abbreviations
- ASB-16:
-
amidosulfobetaine-16
- BH3:
-
Bcl-2 homology 3
- CuPhe:
-
copper(II)(1,10-phenanthroline)3
- DMEM:
-
Dulbecco’s Modified Eagle Medium
- DTT:
-
dithiothreitol
- FCS:
-
fetal calf serum
- GFP:
-
green-fluorescent protein
- HA:
-
haemagglutinin
- hBak:
-
human Bak
- IEF:
-
isoelectric focusing
- IRES:
-
internal ribosome-entry site
- MEFs:
-
mouse embryonic fibroblasts
- pI:
-
isoelectric point
- PKA:
-
protein kinase A
- pTyr:
-
phosphotyrosine
- PVDF:
-
polyvinylidene fluoride
- tBid:
-
truncated Bid
- wtBak:
-
wild-type Bak
References
Westphal D, Dewson G, Czabotar PE, Kluck RM . Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta 2011; 1813: 521–531.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Lindsten T, Ross AJ, King A, Zong W, Rathmell JC, Shiels HA et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389–1399.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Cory S, Huang DC, Adams JM . The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 2003; 22: 8590–8607.
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ . Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002; 2: 183–192.
Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 2011; 44: 517–531.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17: 393–403.
Dai H, Smith A, Meng XW, Schneider PA, Pang YP, Kaufmann SH . Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol 2011; 194: 39–48.
Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM et al. To trigger apoptosis Bak exposes its BH3 domain and homo-dimerizes via BH3:grooove interactions. Mol Cell 2008; 30: 369–380.
Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T et al. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ 2012; 19: 661–670.
Oh KJ, Singh P, Lee K, Foss K, Lee S, Park M et al. Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3-BH3 contact interface in BAK homo-oligomers. J Biol Chem 2010; 285: 28924–28937.
Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM . Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol Cell 2009; 36: 696–703.
Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J et al. A stapled BID BH3 helix directly binds and activates Bax. Mol Cell 2006; 24: 199–210.
Kutuk O, Letai A . Regulation of Bcl-2 family proteins by posttranslational modifications. Curr Mol Med 2008; 8: 102–118.
Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 2007; 27: 4006–4017.
Domina AM, Smith JH, Craig RW . Myeloid cell leukemia 1 is phosphorylated through two distinct pathways, one associated with extracellular signal-regulated kinase activation and the other with G2/M accumulation or protein phosphatase 1/2A inhibition. J Biol Chem 2000; 275: 21688–21694.
Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J et al. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem 2002; 277: 43730–43734.
Moujalled D, Weston R, Anderton H, Ninnis R, Goel P, Coley A et al. Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-only protein Bim. EMBO Rep 2011; 12: 77–83.
Ley R, Ewings KE, Hadfield K, Howes E, Balmanno K, Cook SJ . Extracellular signal-regulated kinases 1/2 are serum-stimulated ‘Bim(EL) kinases’ that bind to the BH3-only protein Bim(EL) causing its phosphorylation and turnover. J Biol Chem 2004; 279: 8837–8847.
Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM et al. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol 1999; 144: 903–914.
Fox J, Azad A, Ismail F, Storey A . ‘Licensed to kill’: tyrosine dephosphorylation and Bak activation. Cell Cycle 2011; 10: 598–603.
Fox JL, Ismail F, Azad A, Ternette N, Leverrier S, Edelmann MJ et al. Tyrosine dephosphorylation is required for Bak activation in apoptosis. EMBO J 2010; 29: 3853–3868.
Anderson JC, Peck SC . A simple and rapid technique for detecting protein phosphorylation using one-dimensional isoelectric focusing gels and immunoblot analysis. Plant J 2008; 55: 881–885.
Gianazza E . Isoelectric focusing as a tool for the investigation of post-translational processing and chemical modifications of proteins. J Chromatogr A 1995; 705: 67–87.
Rabilloud T, Chevallet M, Luche S, Lelong C . Two-dimensional gel electrophoresis in proteomics: past, present and future. J Proteomics 2010; 73: 2064–2077.
Fiol CJ, Williams JS, Chou CH, Wang QM, Roach PJ, Andrisani OM . A secondary phosphorylation of CREB341 at Ser129 is required for the cAMP-mediated control of gene expression. A role for glycogen synthase kinase-3 in the control of gene expression. J Biol Chem 1994; 269: 32187–32193.
Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM . Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Natl Acad Sci USA 2009; 106: 1760–1765.
Lessene G, Czabotar PE, Colman PM . BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov 2008; 7: 989–1000.
D’Alessio M, De Nicola M, Coppola S, Gualandi G, Pugliese L, Cerella C et al. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J 2005; 19: 1504–1506.
Ferrer PE, Frederick P, Gulbis JM, Dewson G, Kluck RM . Translocation of a Bak C-terminus mutant from cytosol to mitochondria to mediate cytochrome C release: implications for Bak and Bax apoptotic function. PLoS One 2012; 7: e31510.
Balmanno K, Cook SJ . Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ 2009; 16: 368–377.
Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak.[see comment]. Science 2007; 315: 856–859.
Gordon JA . Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol 1991; 201: 477–482.
Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B et al. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem 1997; 272: 843–851.
Moldoveanu T, Liu Q, Tocilj A, Watson MH, Shore G, Gehring K . The x-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 2006; 24: 677–688.
Acknowledgements
We thank Hamsa Puthalaketh, Sandra Nicholson and Jarrod Sandow for advice and reagents related to protein phosphorylation, Nicole Church and Thomas Nebl for advice on 1D-IEF, and Stephanie Fennell for technical assistant. The work was supported by grants from the National Health and Medical Research Council of Australia (no.575559, no.1016701 and no.637335), and the Association for International Cancer Research (no. 10–230), and operational infrastructure grants through the Victorian State Government Operational Intrastructure Support and the Australian Government NHMRC IRIISS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Raschellá.
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tran, V., Bartolo, R., Westphal, D. et al. Bak apoptotic function is not directly regulated by phosphorylation. Cell Death Dis 4, e452 (2013). https://doi.org/10.1038/cddis.2012.191
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.191
Keywords
This article is cited by
-
TRPM7 overexpression enhances the cancer stem cell-like and metastatic phenotypes of lung cancer through modulation of the Hsp90α/uPA/MMP2 signaling pathway
BMC Cancer (2018)
-
Mcl-1 and Bcl-xL sequestration of Bak confers differential resistance to BH3-only proteins
Cell Death & Differentiation (2018)
-
Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains
Cell Death & Differentiation (2015)
-
Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax
Cell Death & Disease (2015)
-
Dissociation of Bak α1 helix from the core and latch domains is required for apoptosis
Nature Communications (2015)

{kind=link}
{kind=link}
{kind=link}